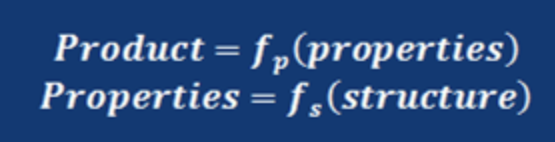| Home | 化学 | HSP | 情報化学+教育 | PirikaClub | Misc. |
2020.10.22
非常勤講師:山本博志
Joback先生とは2014年以来の付き合いだ。
私は20年以上前から物性推算と逆設計をやっていたので、当然、ご高名なJoback先生の事は知っていた。私のWebページでもJoback法の物性推算式は取り上げている。
たまたま、2014年に分離技術会にJoack先生が呼ばれて講演会をする際に、私も一緒に発表する機会があった。
そこで、Joback法を拡張する、Dynamic Groups Contribution(動的原子団寄与法)の話をした。
それ以来、先生が日本に来るたびにお会いしている。2017年に日本にいらした時には、奥様と一緒に拙宅にお招きして、夜中の3−4時まで色々なことを議論した。
Joback先生の奥様は日本人で、年1回ぐらいは帰省している。先生は僕と同じ年なので、大学時代のコンピュータの状況などは近いものがある。
議論をしていると非常に楽しい。
二人が議論を始めると、奥さん連は「なんて似た者の二人だ」と呆れ返る。
今年もInchem Tokyo 2019に来日するのに合わせて打ち合わせをすることにした。

Joback先生は修士の時にJoback物性推算法,博士の時に逆設計法を確立したMIT(マサチューセッツ工科大学)の秀才だ。
Joback法は様々なプロセス・シミュレータに搭載されている。
Scifinderのようなデータベースでも、実測値のない化合物の所にJoback Method推算値が書かれていることがある。
だからJoback先生のお名前は聞いたことがある方も多いと思う。
Molecular Knowledge Sysytemsという会社を設立して、物性推算用のソフトCraniumと、逆設計用のソフトSynapseを販売している。
Materials Informatics(MI:マテリアルズ・インフォマティクス )やMaterials Genome(MG:マテリアルズ・ゲノム)とかいうものが、最近巷を賑わしている。
私は20年以上前から、Joback先生は40年近く前から、ずーっとやって来たことだ。
みんなに判る私たちの仕事
簡単に言ってしまえば、Joback先生のHPにあるこの図が全てだ。
材料(分子)物性は様々な物性値(原子団構造)の関数として表すことができる。
様々な物性値は物性推算を行うことで得られる。
でも、素材開発を行う側は、ある要求物性値を満足する構造(構成)の関数を知りたいことが多い。
特に要求物性が複数ある時にには、関数の数が増えて来るので人間の力だけではどんどん大変になる。
先生の奥さんは日本語、英語に堪能であるが、化学のことは堪能では無い。
今回私の大学での授業を元に、私たちの研究のコンセプトを、奥さんにわかるように、次のように説明した。
例えば、爪を光らせるマニュキュアがあります。
それを取り除くリムーバー(除光液)があります。
それらにはこれまでアセトンが溶媒として使われて来ました。
しかし、アセトンの中毒性の問題から、最近はノン・アセトンタイプのものが増えて来ています。
商品の裏を見るとMEK(メチル・エチル・ケトン)を使っているとか記載があります。
確かにノン・アセトンですが、MEKはアセトンよりもLD50(半数致死量)が小さい毒性の高い溶剤です。
そのような溶剤に変えるのは意味がないですよね。
それではマニュキュア、リムーバーの溶剤にはどのような性質が必要でしょうか?
爪を光らせるために塗っているのはニトロセルロースというポリマー(高分子)です。
溶剤というからには、1.溶解性が大事になります。
刷毛で塗るので、2.粘度が適当な範囲に入っている必要があります。
塗った後に乾いてくれないと困るので、3.室温での蒸気圧が必要になります。
もちろん、4.毒性や,5.引火性なども重要です。
挙げればキリがありませんが、アセトンを他の溶剤に切り替えようとした場合には、ある溶媒の様々な物性を集めなくてはなりません。
全ての物性値がカタログに書いてあればデータベースを検索すれば欲しい溶剤が出て来るのでしょうが、残念ながらそうしたデータベースはありません。
そこで、無い物性値はCraniumなどの物性推算ソフトウエアーで推算することになります。
PubChemというデータベースには300万以上の化合物が登録されています。
そうした化合物全てについて、1-5の物性値を計算して所定の範囲に入った化合物を抜き出して来る?
残念ながら、そのような設計は難しいのです。
実際には除光液などは複数の溶媒の混合液として使われます。
複数の溶媒を選んで、どのような比率で使うか?
それを全て評価して候補組成とすることはできないのです。もちろん、工業的に利用可能+費用が適当な溶剤はすごく限られます。それでも組み合わせの数は膨大です。
そこで、Synapseなどの逆設計ソフトウエアーが必要になります。要求特性を入力して探索!とすると、構成を探し出してくれます。
そんな説明をしました。
皆さんお分かりになりましたか? 先生の奥さんは分かってくれました。
物性推算ー逆設計とマテリアルズ・インフォマティクス の違い
実は違いなど無い。
私は2000年に触媒学会の招待公演で、酸化物ガラスの組成設計の話をした。
その時には物性推算と逆設計と言っていた。(発表タイトル「ニューラルネットワークを用いた酸化物ガラスの物性推算と遺伝的アルゴリズムを用いた組成決定システムの開発」)
それはChemo-Informaticsという化学情報学の分野に属する技術だ。
2010年に論文、「ガラスの組成設計技術:Design of Glass Compositions」を書いた時も、物性推算と逆設計と言う言い方をしている。
2011年にアメリカでMaterials Genome計画がスタートしてから、材料ゲノムという言い方が流行りだした。
私が2000年に発表した遺伝的アルゴリズムという手法(自分のオリジナルでもなんでも無い。よく使われる方法)はガラスの組成を遺伝子(ゲノム)に見立てて、適者生存の法則で逆設計をする。
材料の構成を遺伝子(ゲノム)に見立てるので、マテリアル・ゲノムと呼んでいるのだろう。
その後、Materials Informaticsという言い方が流行り始めた。
最適解探索に遺伝的アルゴリズムだけでなく、informatics(情報学)全般を利用しているのだろう。
言葉は変わっても、物性を予測して、逆設計をする。
結果を予測式にフィードバックして予測性能を上げ、逆設計を繰り返し、高速に欲しい答えにたどり着くということを目指しているだけだ。
ただし、私とかJoback先生のやっている物性推算と逆設計は最近のMI,MGとはちょっと違うクラッシックなものだ。
Joback先生は化学工学出身なだけあって、旧来の化学工学的な経験式を多用する。
私は一時期量子化学にはまり、10年ちょっと前に化学工学で博士号をとった事もあり、そのハイブリッドを多用する。
この経験式をベースにしているところが私たちの1番の強みだ。
熱力学的な物性値を扱う時には臨界定数から対応状態原理を使う。
溶解度を扱う時にはハンセンの溶解度パラメータという経験式を使う。
混合物を扱う時にはASOG法という活量推算式を使う。
必要であれば量子化学計算の結果も使う。
経験式というのは、実在の現象を(理論はよくわからなくても)数式化したものだ。
理論ができてコンピュータが早くなれば、経験式などいらなくなる。
そんな時代も将来くるかもしれない。
しかし、現状ではJobackの臨界温度推算法の精度は、分子動力学法(MD)で求めるものより圧倒的に精度が高い。
フローリー・ハギンスのΧパラメータは、ハンセンの溶解度パラメータから求める方が圧倒的に便利だ。
MO,MDだけをベースにするデータ・サイエンティストにはできない芸当だ。
やっていることは同じだが、やり方が違う。
活量推算式ASOGに愛を入れよう
色々なものが混合された時には複雑なことが起こる。
純物質系の物性推算に関しては、経験式、理論式どちらも随分と進歩した。
有機系であれば、構成する原子は、炭素、水素、酸素、窒素、燐、硫黄、ハロゲンを網羅すれば、98%ぐらいカバーするだろう。
こうした原子に関しては1990年ぐらいには半経験的分子軌道法MOPACでもパラメータが決まり分子設計に使われて来た。
更に広い原子に使うのであれば、ガウシアンなどを使わなくてはならない。
有機金属触媒設計などはそちらに進んで行った。
では、普通の98%の化合物を計算するのに、MOPAC程度でいいか、ガウシアンが必要か、は議論が分かれるところだろう。
先ほどのマニュキュア用の溶媒設計でアセトンをMEKに変えるのはナンセンスなのは自明として、他の溶媒システムに変えることを考えよう。
1.ニトロセルロースの溶解性
2.溶液粘度
3.室温での蒸気圧
4.毒性
5.引火性
この中で分子軌道法の計算結果から判る物性値はどれだろう?
分子間力を厳密に計算できて、分子動力学(MD)が進歩すれば、溶解性、粘度、蒸気圧、引火性は判るかもしれない。
毒性に関しては未だに成功していない。
でも、ガウシアンの基底関数をどんなに高くしても、沸点を±5℃で予測することは困難だ。
沸点近傍では1℃違えば、蒸気圧は30mmHg異なる。
まー現実的には、分子の安定性の尺度、生成熱を引火点に結びつける計算ぐらいが関の山だろう。
その時に、MOPACで計算される生成熱とガウシアンで計算される生成熱とどちらを使うのが良いだろうか? どの道、物性値ズバリを表す指標が無いのなら、
Propertie=f(Descriptors)
という関数fで定義するしかない。
生成熱は識別子の1つとして使える。
しかし塩素の入った化合物は、塩素がラジカルとして解離する場合、燃焼のクエンチ剤として作用する。そして引火点を無くしてしまうことがある。
CF3などの官能基もそういう作用がある。
パーフルオロのアルカンは引火点が無い。
それは分子が安定だからという言い方もできる。
しかし、パーフルオロのカルボン酸エステルは、アルキル基の長さがどんなに長くてもエステルの部分が燃えてしまうので引火点を持つ。
また、ある温度での蒸気の量は引火点と高い相関がある。
イオン液体などは蒸気圧が無いので引火しない。
つまり、生成熱の計算精度をどんなに高くしても、引火点の推算精度は高くならない。
そこでJoback先生や私は、生成熱の推算に関し、MOPACの計算程度で十分、更にはJoback法、原子団寄与法で十分という立場になる。
きちんとした原子団を定義して、Hansenの溶解度パラメータを推算して溶解性や溶液粘度を推算して、対応状態原理を使って蒸気圧を推算して、必要な物性値を得ていく。
更に混合溶媒になっていくと、原子団寄与法と、MOPAC、ガウシアンの理論計算の差は広がっていく。
化学工学には経験的なMixing Ruleはたくさんある。
それらを理論的な方法だけで再構築するのは、まだまだ大変なことだ。
COSMO-RSのような方法もあると言うかもしれない。
これを使うと気液平衡などを精度高く推算できたと言う例も発表されている。
しかしよく聞くと蒸気圧式は実験値のAntoine方程式を使っていると言う。
それでは、Antoine定数の無い化合物をどうやって計算するのか?といえば、経験式を使わざるを得ない。
Joback先生や私も経験式の上にどっかり胡座をかいてのんびりしているわけでは無い。
(アカデミックは無料で使える)MOPACの計算結果は最大限に利用するし、経験式の改良も強力に推し進めている。
今回の話し合いの中で、AiSOGをCraniumやSynapseに搭載していこうと言う話になった。
ASOG法と言うのは、Analytical Solution of Groupsの略だ。
原子団ごとに割り振った、局所活量係数パラメータを用いて混合物のWilson活量係数を推算する。
それを用いて気液平衡や液液平衡などが推算できる。
このASOG法は日大の栃木先生が50年前から開発を続けている方法だ。
私は栃木先生のところで博士号を頂いているし、Joback先生を分離技術会の講演会にお招きしたのも栃木先生で、浅からぬ縁がある。
ASOG法の精度はUNIFAC法に引けを取らない素晴らしい精度を持っている。
問題点をあげるなら、原子団のペアでパラメータが決まっていないものが沢山あることと、パラメータを決めるのが難しいことだろう。
詳しい方法はここでは割愛する(pirikaのWebページを参照して頂きたい)が、この問題に関して、AI技術を導入して適用範囲を広げる取り組みをして来た。
AI-ASOG、縮めて、AiSOGプロジェクトだ。
これをJoback先生の製品に搭載する予定にしている。
経験式であれ活量係数の教えてくれることは色々ある。
活量係数次第で、液体の組成と気体の組成は大きく異なる。
気相の不燃成分の割合が高くなれば引火点を持たなくなる。
生体ポリマーのアミド基やチオール基と医薬品の、局所活量係数がわかれば、何故その医薬品がある特定部位で作用するのかがわかり、ドラッグ・デリバリー・システム(DDS)の開発に繋がる。
純理論計算でなく、既に確立しているクラッシックな技術の延長線で。ベースになるのが、愛(i)を入れたASOG, AiSOGだ。
所変われば品変わる
これだけ長いこと、物性推算と逆設計をやっていると色々思うことがある。
本質的にやりたいのは逆設計である。
逆設計を精度高く行いたい場合に、物性推算の精度が高くなることは必須であろうか?
私とJoback先生の答えは否である。
先にも書いたが、引火点を推算するのに生成熱の情報は大事である。
それを原子団寄与法から求めるのと、ガウシアンから求めるのでは、ガウシアンの方が精度は高いだろう。
でも、逆設計する度に、化合物や混合物をガウシアンで計算しなくては逆設計できないのであれば、いくら計算機のスピードが早くなっても大変なことだろう。
でも、実はスピードの問題では無い。
量子コンピュータが実現してもダメだろう。
原子団寄与法であってもニューラルネットワーク法などを使って推算精度を上げることはできる。
PLSやPCA法など様々なケモ・インフォマティクス に使われている精度の高い推算方法も進歩している。
そうした推算式を使えば十分高速な推算式が構築できる。
ガラスのレンズの色収差をなくすと言う目的なら、屈折率とAbbe数の推算精度は高ければ高いほど良いだろう。
材料が手に入ったら、コンピュータの逆設計で、厚みや形状を最適してガラスレンズが得られる。
しかし、薬化合物のLUMOが薬効に効くからといって、-1.2435±0.0005とガウシアンで出そうが、-1.24±0.05とMOAPCで出そうが、逆設計には何の役にも立たない。
LUMOを変えるためには化合物の構造を変えなくてはならず、それを変えれば、他の物性値は皆変わってしまう。
精度が高かろうが低かろうが関係ない。
分子軌道法が逆設計に使えないと言う所以である。
ガラスの物性なら組成を微妙に変えることはできるが、薬設計はそれができない。
ポリマーを使ったプラスティック・レンズならどうだろう?
低分子と異なり高分子の物性はある範囲でしか求まらない。
屈折率は1.67-1.69の間と言う具合にだ。
様々な会社が様々な開始剤、連鎖移動剤を使って、いろいろな重合方法で重合し、分子量分布も異なり、精製方法も、安定剤も異なる。
同じPMMAといっても物性値に範囲が出てしまう。
そうしたポリマーの物性推算の精度が高くても意味はない。
目標とするスペックに対して、添加剤やプロセス条件を逆設計をする。
国家プロジェクトでやるような、よっぽど特殊な材料はプロに任せて、我々は目的に合った逆設計方法を模索する。
では、推算式の精度向上に興味がないかといえば、そんな事はない。
材料開発のキモは逆設計だと信じている。
逆設計のキモは取りこぼしのない事だと思っている。
現状のニューラルネットワーク法は、ビッグデータがない限り、過学習を起こし、すっぽ抜ける点を多数持っている。
学習した点近傍で大きく答えが変化し、しかもそれが多次元だから理解が難しい。
かえって重回帰分析ぐらいの方が取りこぼしが少なくなる。
しかし、重回帰分析では非線形現象は扱えない。
他社や人間が気がつかないような非線形現象をうまく使って、他にマネのできない材料を設計したいと言う目的には叶わない。
そこで私たちの開発しているのはJoback-Yamamoto Dynamic Groups Contributionという推算法だ。
逆設計で取りこぼしをしない事を主眼において最適化した非線形の推算法だ。
詳しいことは、そのうちpirikaで説明しよう。これもJoback先生の製品に搭載する事を考えている。
現場の知恵
現場の方々は様々な知恵を持っておられる。
こういう時には、この成分をちょっと加えれば良いなど、理論的には説明できない精緻な経験式と言える。
例えばガラスの物性を推算するのに、Appen式というものがある。
係数が範囲でしか定められていないものもあるが、組成を入れれば様々な物性を推算できる。
範囲でしか求まらないものは、例えばホウ素のように、入れた量に対して、BO3の三角形であったり、BO4の四面体構造だったりと構造が変わるので物性も非線形に変わる。
しかし、そうした異常性以外に、混合アルカリ効果などの複合化することによる異常現象も沢山ある。
スズの異常性をうまく使って線形では出せない素晴らしい複合酸化物を設計して特許を書いている人もいれば、ストロンチウムを使ってうまくやっている人もいる。
物性値がそもそもAppen式の答えと同じなら、新規性は無いので特許にはならない。
そうした特許を集めてニューラルネットワークに学ばせると、現場の知恵をデジタル化できる。
合金設計や触媒設計も同様だろう。
Joback先生は有機系が強い。私は無機系も強い。色々楽しそうなことができそうだ。
でもコンセプトはいつもと同じ、物性推算と逆設計だ。
有機物であろうが、無機物であろうが同じだ。
それにしたって、大事なのは現場の知恵だ。それを捨て去ってMIだ何だと騒いでも上手くいかないだろう。
オープン・コラボレーションと下請け
最近、オープン・コラボレーションとかいう言葉が流行っている。
全てを自前主義でやるのでは無く、不得意なところを、それが得意なところへ任せてしまおうという発想らしい。
そういった言葉はどうも独り歩きをしているようにも思える。
車と電気産業のように補完しあう関係に使うならわかる。
ところが、MIはできる人材もいないし、不得意だからできる所に任せてしまおうという時にも使われる。
それは、オープン・コラボレーションでは無く、単なる下請けだ。
通常、下請けというのは、簡単な仕事(例えば掃除)で、本体の優秀な、高級とりの人材にやらせるまでも無い事を頼む時に使う。
長い不況を経て、労働者の非正規雇用の割合は30%を超えた。
理系の大学院を卒業しても非正規で働いている方もおられる。
下手をすると、仕事を頼んでいる人よりも高学歴な事もある。
高学歴な研究者を安い給料でこき使える事を覚えてしまうと、そこの所を誤解してしまう。
本体でできない高度なMIを外注するなら、本体よりも高コストになるはずだ。
事実、電機メーカーは優秀な人材を取るために、年収1000万超も提示し始めた。
しかし、それをやると中堅の社員とのバランスが悪くなる。
そうした時に使うのがオープン・コラボレーションだと思う。
大学発のベンチャーで"MIやります”なんていう会社が雨後の筍のように現れている。
学生という安い労働力を持っている強みだろう。
自社のプロの研究者のレベルが、素人の学生のレベルに劣るなら、そうした所を使うのも良いだろう。
価格破壊が起きている今がチャンスだ。
Joback先生も私も大学で教えてはいるが、研究室を持っているわけでは無いので安い学生の労働力は持っていない。
従って下請けはできない。
そうした環境で先生と考えている仕組みは、ある汎用基盤は我々が作る。
そこに会社独自のデータを入れると、会社の目的に特化したシステムに成長していくシステムだ。
AiSOGで言えば、局所活量係数の重要度は会社によって異なる。
自社が使う原子団だけが高精度であれば良い。
しかし特注ソフトは高価になる。
汎用ソフトは数が売れるので安価になる。
独自データは外には出したく無い。
汎用ソフトを自社用に進化させるには、(MIがわからなくても)自社のプロが役に立つ。
そんな仕組みを導入しようとしている。1%の会社が理解してくれれば良い。
大学での講義資料を融通しあったりしながら、楽しく付き合っていく間柄だ。
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください) メールの件名は[pirika]で始めてください。