Hansen Solubility Parameters in Practice (HSPiP) e-Book Contents
(How to buy HSPiP)
Chapter 16, HSP and Diffusion
The good news is that although the
calculations of diffusivity are rather hard to implement, the theory of what is
going on is remarkably simple. A diffusion modeller is included within the HSPiP
package so you can explore the theory without having to worry about its
detailed implementation. In what follows we are referring only to solvent
diffusion under constant temperature and are not discussing diffusion driven by
other gradients such as temperature.
For those who just want a quick method to
estimate diffusion coefficients, the official Nordtest Poly 188 found at http://www.nordicinnovation.net/nordtestfiler/poly188.pdf
gives a well-validated methodology that is a simplified version of what is
found in this chapter. It is no coincidence that Hansen was one of the authors
of the Poly 188 test.
Contrary to popular mythology, there is
only one type of diffusion that is important for polymers in normal practice.
If you do practical experiments you might be surprised by this statement
because it’s rather obvious that there are at least two distinctly different
situations of interest: absorption (solvent going into a polymer) and
desorption (solvent coming out of a polymer). This is obvious to anyone who has
done absorption/desorption experiments because absorption is generally much
faster than desorption. Those of you who know more about the topic will also be
aware that some diffusion rates are simple (“Fickian” diffusion) and some are
complex (e.g. “Type II” diffusion).
The response to this apparent complexity is
to repeat that there is only one type of diffusion based on just one simple
equation, the diffusion equation (sometimes called Fick’s second law). The
apparent differences between all the different types reflect the fact that
different factors involved in this equation are more or less important
depending on circumstances. The reason to stress this simple unity is that each
of the different factors is rather easy to understand. It is therefore rather
easy to work out which factors are required to sort out what is happening in
just about any diffusion behaviour. Armed with HSP of solvent and polymer, with
some knowledge of molar volume (integral to HSP) and molecular shape, and with
some understanding of whether (and when!) a polymer is highly crystalline,
semi-crystalline or elastomeric (often reflected in the HSP Radius), you can
readily calculate diffusion behaviour. As discussed below, the diffusion rate
increases in the order highly crystalline < semi-crystalline < elastomeric as governed by Factor 5.
These categories are gross simplifications only to be used as rules of thumb
– there are certainly cases of crystalline polymers with higher diffusion
rates than semi-crystalline. But these rules are a good starting point. We
provide some more rules of thumb on diffusion rates in the text and in the HSPiP
modeller.
Consider the following factors, one by one.
Factor
1. This is the Mass
Transfer Coefficient, h. A large h means that if you have a well-stirred liquid
in contact with the polymer surface then you have no shortage of molecules to
be able to diffuse into the polymer. If, on the other hand the polymer surface
is in contact with still air, then a boundary layer builds up and solvent
trying to escape from the polymer will see a high concentration of vapour which
reduces the gradient for diffusion and therefore slows down the rate, so h is
small. One reason for there being a difference in absorption and desorption in
this case is the simple and obvious fact that the Mass Transfer Coefficient
from 100% liquid in absorption is much higher than that into a boundary layer
of air (saturated with vapour) in desorption. But there also can be times when
mass transfer into the polymer from a liquid is limited (e.g. poor stirring of
an inert carrier liquid such as water, with fast absorption of a solute). In
all of these cases there is rapid diffusion of the chemical within the polymer
and some problem with mass transport more or less exterior to the polymer.
Another factor which can cause large reductions
in the mass transfer coefficient is the formation of a highly-crystalline skin
at the surface. The absorbing molecule simply cannot find any access point into
the bulk (less-crystalline) polymer. The reduction in mass transfer coefficient
is larger for larger and/or more complex molecules. Above a certain size the
mass transfer coefficient becomes zero – there is no penetration into the
bulk. The most extreme example of this is when two polymers, even well-matched
in terms of HSP, simply cannot interpenetrate. What is surprising is that even
some simple molecules (e.g. a single benzene ring or a modestly branched
structure) can have a mass transfer coefficient close to zero for some polymers
such as those with high crystallinity or other forms of close-packing such as
are found in the Topas polymers. Chapter 16 of the Handbook discusses some of these issues in greater detail.
A good method for checking if diffusion is
limited by mass transfer is to carry out the experiments on samples of
different thicknesses – the thicker the sample, the less relevant the
mass transfer effect becomes – as you can readily determine using the
diffusion modeller.
Given that most of us don’t know what the
Mass Transfer Coefficient is for our systems, the modeller uses the Hansen B
value. This is the ratio of the diffusion resistance to the surface resistance.
With D0 as the diffusion coefficient at the lowest concentration
encountered, and for a free film sample of thickness L, the diffusion
resistance is (L/D0). Dividing this by the surface resistance (1/h)
gives B. Thus,
B= hL/D0
A high B (>10 for a constant diffusion
coefficient) means essentially no significant limitation by mass transfer.
Factor
2. This is the local
saturated concentration of the liquid in the polymer right at the surface
during absorption. For RED less than 1 for a correlation based on “good” or
“bad” solubility, this concentration will be very high since the solvent can in
principle completely dissolve the polymer. It is very difficult to assign an
initial given surface concentration, but it is probably in the range of 50-70%,
because at still higher concentrations, the issue is not one of diffusion of
solvent into the sample but diffusion of the sample into the solvent. For RED
larger than 1, the larger the value, the lower the local saturated
concentration and therefore the slower the absorption rate. This is the reason
that HSP is so important for understanding diffusion. In the modeller a simple
algorithm has been used to illustrate this so you can compare overall diffusion
rates as the RED changes. The algorithm is for illustrative purposes only
– it’s up to you to specify the surface concentration in any specific
scenario.
Once the molecule is inside the polymer, as
long as it is within its solubility limit (we’ll explain this in a moment), HSP
play no further role. The rates of diffusion of a low RED and high RED solvent
of similar molar volume and shape are the same. You might be surprised that in
an HSP book it is claimed that HSP are not important for diffusion inside the
polymer, i.e. the diffusion coefficient at a given concentration. The
experimental data have confirmed this fact many times. This also means that HSP
play no part in classic desorption experiments to air. Naturally the desorption
from one polymer to another (migration) does
depend on the HSP of the second polymer as a large HSP mismatch would mean, as
in absorption, a low surface concentration in that polymer.
Although Factor 2 is about absorption, it’s
a good point to discuss why desorption takes so much longer than absorption. It
has been shown that the diffusion coefficient increases exponentially with the
concentration of the solvent. For rigid polymers this increase is a factor of
about 10 for each 3%v increase in solvent concentration. For flexible polymers
the increase is a factor of 10 for about 15%v increase in solvent
concentration. Whereas during the whole time of the absorption process, the
solvent is largely diffusing in at concentrations approaching the maximum
(surface equilibrium) concentration, and certainly much higher than the lowest
concentration, in desorption most of the solvent diffuses out at much lower
(and falling) concentrations than the initial one. In desorption the concentration
at the surface is low (zero) so the process is largely controlled at or near
the exit surface since the diffusion coefficient here is so low.
Before going to Factor 3, let us clarify
this statement that “as long as it is within its solubility limit, HSP play no
further role.” If you dip some polymers (e.g. epoxies or polypropylene) into
hot water, the solubility is increased sufficiently for water to diffuse in
(the δH of water falls off rapidly with temperature, boosting its solubility).
If you then cool the sample, the water becomes insoluble in the polymer. The
individual water molecules can still diffuse (diffusion coefficient is
independent of HSP) but when they meet each other, they phase separate from the
polymer. This is the classic case of water blisters. If you hot-dip/cool a
number of times you get more and more water into the polymer, but each time you
cool, the water phase separates out into bigger and bigger blisters. The
blisters are very persistent. That is because each blister is a new diffusion
problem from one phase (the water blister) into the other (the polymer). Given
that there is a large HSP mismatch, the surface concentration at the
blister/polymer interface is low so the rate of diffusion is low. Those
blisters can be very persistent. So now you can see why it’s important to
qualify the statement that HSP have no effect on diffusion once the molecule is
inside the polymer. See p141 of the Handbook
or C.M. Hansen, New Developments in
Corrosion and Blister Formation in Coatings, Progress in Organic Coatings,
26, 113-120, 1995 for further details. Incidentally,
a beautiful demonstration of HSP co-solvent effects is provided by the
well-known fact that glycol ethers in coatings can produce blisters under
aggressive thermal/water cycling tests. The ethers remain in the coating and
during the hot/wet part of the cycle the combined glycol ether/water HSP is a
sufficient match to the hot polymer to allow the water to enter. On cooling the
blisters start to form. The same coatings without the glycol ethers have no
blistering because the HSP distance of the water is too great, even at the higher
temperatures.
Factor
3. This is the molar volume. The larger the molar
volume, that is, the size of the molecule, the smaller the diffusion constant.
This is a generalisation that is modified by Factor 4, and therefore it must be
used as a guideline rather than a hard fact. In the modeller the rule of thumb
(based on the rather small number of studies in the literature) is used that
the log of the diffusion constant is proportional to the molar volume. The
constant of proportionality changes strongly from rigid to flexible polymers.
For rigid polymers there is a very strong dependence, so a doubling of molar
volume can result in a 10 to 100-fold reduction in diffusion constant. For
flexible polymers the dependence is weak – a doubling of molar volume may
merely halves the diffusion constant.
Factor
4. This is the molecular shape. A linear, flexible
molecule can easily wiggle through a polymer. A rigid (aromatic) molecule or a
highly branched molecule takes much longer to find a space (or “free volume”
– see Factor 5) in which to wiggle. The rule of thumb section below gives
some examples of the effects of Factors 3 and 4. A well-known table of solvent
diffusion rates from Shell suggests that the combined effects of molar volume
and molecular shape result in a diffusivity order from faster to slower of:
Methanol, Acetone, MEK, Ethyl Acetate, n-Heptane, n-Butyl Acetate, Benzene,
1,4-Dioxane, Toluene, MIBK, i-Butyl Acetate, 2,4-Dimethyl Pentane, Cyclohexane,
Diacetone Alcohol, Pentoxone, Methyl Cyclohexane, Cyclohexanone, Methyl
Cyclohexanone. This table was derived using the technique originally developed
by Hansen when he created the data of Fig 1 of the chapter on glove data.
Factor
5. Informally we can say that Factor 5 is whether
the polymer is rigid (slow diffusion), flexible (faster diffusion), or a
quasi-solution – e.g. an elastomer where the polymer is held together by
just a few crosslinks (still faster diffusion). More technically we can talk
about polymer “free volume” where the rigid polymer is below its glass
transition temperature (Tg) and therefore has little main-chain segmental
motion and the flexible polymer is either naturally above its Tg or has large
free volume for main-chain segmental motion thanks to all the solvent. It is
Factor 5 which causes most of the confusion about diffusion science. The more
free volume there is, (or, equivalently, the lower the glass transition
temperatures), the faster will be the rate of diffusion. The simple view of all
this is that when the solvent enters a polymer it starts to plasticize it and
diffusion is faster. As more solvent diffuses into a polymer the diffusion rate
can increase by factors of 100’s, 1000’s and even millions (the solvent itself
is increasing the polymer free volume) so the diffusion looks more complex.
This “concentration dependent” diffusion is not some sort of special case or
special phenomenon – it is the general rule for polymers. Sometimes it
looks “special” because the concentration dependence is so large – but
this is only because the polymer happens to be rigid and therefore susceptible
to a large increase in diffusivity. This simple view needs to be treated with
caution. Some crystalline regions are so impenetrable to solvents that they act
as permanent blocks so there is little increase in diffusion coefficient. Some
“amorphous” polymers are in fact highly rigid so show a large increase in
diffusion rate whilst others are highly flexible and therefore show a small
increase. The following figure is for diffusion of chlorobenzene in
polyvinylacetate.
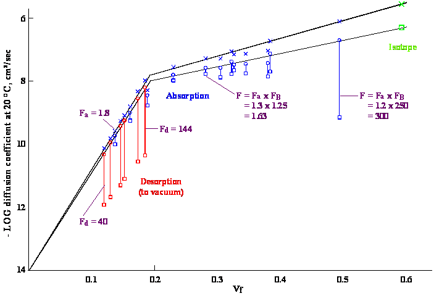
Figure 1‑1 Diffusion coefficients
for chlorobenzene in poly(vinyl acetate) at 23°C measured by absorption and
desorption experiments in a quartz balance apparatus as well as with an isotope
technique. vf is the volume fraction. The upper curve in the figure
is for diffusion coefficients based on total film thickness. The lower curve is
for diffusion coefficients based on dry film thickness as used in the modeller.
It should be noted that the lower curve varies more and more from the upper one
as the concentration of solvent increases. A self-diffusion coefficient for a
liquid (vf = 1.0 in the figure) is a fictitious quantity on the lower curve,
although it is used to define the diffusion coefficient in the solvent rich
regime.
The data in the figure are the result of
combination of absorption and desorption experiments supplemented by isotope
experiments to give a unified view of concentration dependent diffusion in
polymers. In every measurement the observed diffusion coefficient was initially
considered as a constant that must be adjusted to the change in concentration
within the film during the whole process. Solutions to the diffusion equation
with different concentration dependencies were generated and compared with that
for a constant diffusion coefficient to develop these “F” factors. The
apparent, constant diffusion coefficients are given by squares in the figure
with the corrected values being given by circles. The adjustments are for
absorption, desorption, or surface effects as indicated by the subscripts a, d,
and B. Desorption experiments take place largely at local concentrations within
the film that are much lower than the initial concentration that is ascribed to
the experiment. These adjustments are much larger for desorption than for
absorption. The correction for surface effects in the absorption experiment at
vf = 0.5 is a multiplier of 250. Such experiments should not
normally be used to measure diffusion coefficients at these intermediate
concentrations. The procedure used for these adjustments is described in more
detail in the Handbook. The upper
curve is for diffusion coefficients based on the wet film thickness, while the
lower curve is for dry films. It is clear that there are two different regimes,
rigid at lower concentrations, and elastomeric at higher concentrations,
separated by the break at about 0.2 volume fraction of chlorobenzene.
Diffusion coefficients at very high solvent
concentrations are usually best described based on total film thickness rather
than dry film thickness, since the value for the latter at zero polymer
concentration becomes meaningless. A value at 100% liquid is required to define
the diffusion coefficient curve, however, and this value will be somewhat lower
than that found in the literature for self diffusion in the given liquid.
Fortunately, diffusion at very high solvent concentrations is usually so rapid
as to not be a significant effect in the situations of major interest, so
smaller deviations in this region are not important. Whether diffusion is very
rapid or “super-rapid” does not really matter since the process is controlled
by what happens at (much) lower concentrations. Usually the surface
concentrations at equilibrium for absorption or permeation and the start
concentrations for desorption are sufficiently low to allow neglect of this
effect.
The modeller gives you full control over
all these factors. It assumes three regimes that change at two critical solvent
concentrations (which you can choose). Each regime has a diffusion coefficient
which depends on a D0 value (i.e. the value at the lowest
concentration for which this regime is applicable) and an exponential “k” x
concentration term which reflects the increase in diffusion rate. The larger
the k, the larger the increase in diffusivity with concentration:
Drigid = D0r exp(kr x
concentration)
Dflexible=D0f exp(kf
x concentration)
Dsolution=D0s exp(ks
x concentration)
Some
useful data and rules of thumb for Do, cm2/s
In polyvinylacetate at room temperature
(23°C):
|
Liquid |
D0,
cm2/s |
|
Water |
4x10-8 |
|
Methanol |
4.5x10-10 |
|
Ethylene glycol monomethyl ether |
2x10-12 |
|
Chlorobenzene |
1x10-14 |
|
Cyclohexanone |
1x10-15 |
|
Chlorobenzene
concentration %v |
|
|
0.2 |
1x10-8 (changeover from rigid
to flexible-type behaviour) |
|
0.59 |
3x10-6 (changeover from
flexible-type behaviour to solution-type behaviour) |
|
0.76 |
9x10-6 |
|
Pure
solvents (self diffusion unless indicated otherwise): |
|
|
Chlorobenzene (25°C) |
1.7x10-5 |
|
Chlorobenzene (10°C) |
1.3x10-5 |
|
Chlorobenzene (40°C) |
2.0x10-5 |
|
Ethanol (25°C) |
1.2x10-5 |
|
Water (25°C) |
2.3x10-5 |
|
Glycerol in ethanol (25°C) |
0.6x10-5 |
|
In polystyrene |
D0r: |
|
Chloroform |
3x10-13 |
|
In
polyisobutylene |
D0f: |
|
n-Pentane |
2.5x10-9 |
|
Isopentane |
1.2x10-9 |
|
Neopentane |
0.1x10-9 |
Diffusion coefficients above about 10-8 cm2/s
appear to indicate elastomeric behaviour in otherwise amorphous, rigid
polymers, but this value may be lower for true elastomers.
There is an increase in
diffusion coefficient:
For rigid polymers: a factor of about 10 for each additional 3%v
For flexible polymers: a factor of about 10 for each additional 15%v
Special
Cases and Combinations
By breaking down diffusion into these five
factors it becomes easy to disentangle much of the confusion about special
cases such as “Super Case II”. There is really nothing special about these.
Typically what is happening is that the mass transfer limitation (Factor 1) is
interacting with the strong dependency of diffusion on concentration (Factor 5)
in a way that is not intuitively obvious. It’s a useful short-hand to call any
mass transfer effect a “surface resistance” but this term is not very
insightful. A “surface resistance” from poor airflow (desorption) or poor
stirring (absorption) is very different from a “surface resistance” due to a
highly crystalline skin on an injection molded part.
Further confusion arises when tests are
done on very thin parts (or, even, hyper-thin parts when FT-IR measurements are
made on the first few µm of a sample) because then the mass transfer
limitations are proportionally much more significant than on large parts. A
polymer showing an “anomalous” diffusion when tested on thin samples may well
give entirely normal diffusion when tested on a thicker part.
That’s all there is to it. The bad news is
that there is no simple way to calculate each of the five factors. If you are
lucky enough to have reference values of your particular polymer then you are
off to a good start. But the good news is that with the modeller that captures
the essence of each of these factors you can make rapid progress in
understanding whichever system is of particular interest to you. So let’s see
what it can do.
Absorption
and breakthrough
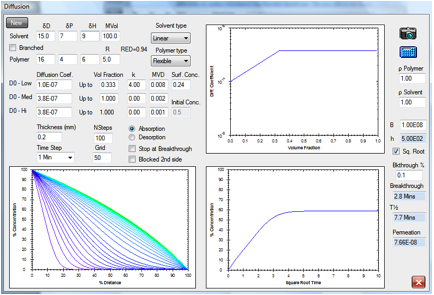
Figure 1‑2 A simple absorption and breakthrough plot
Here we have an elastomer with a
medium-sized, linear solvent. At low concentrations the diffusion rate is 1E-07
and above 0.333 volume fraction the rate becomes constant at 3.8E-07. The
solvent has a RED number of 0.94 and an estimated surface concentration of
0.24. After 2.1min it has broken through (at a 0.1% level) to the other side of
a 0.2mm sample. Shortly after that, the concentration gradient stabilizes to
its final form with the absorption being balanced by the desorption. The
“Square Root” option has been chosen which creates a straight-line in the
increase of % concentration.
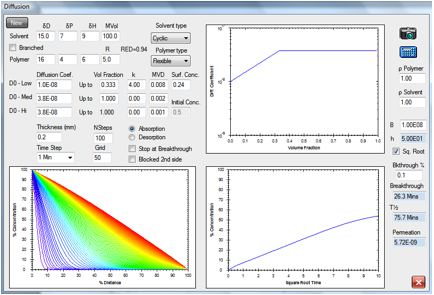
Figure 1‑3 The same absorption but with a slower-diffusing molecule
A cyclic molecule of the same HSP with
twice the molar volume is estimated to have a diffusion rate a factor of 10
slower, so breakthrough time is 26.3min.
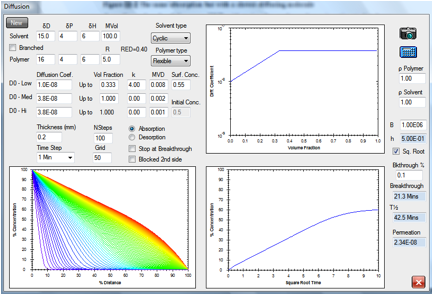
Figure 1‑4 A slower-moving molecule but a lower RED number
A solvent with a RED number of 0.4 but the
same cyclic structure and molar volume is estimated to breakthrough in 21.3min
simply because the surface concentration is estimated to be higher at 0.55.
Desorption
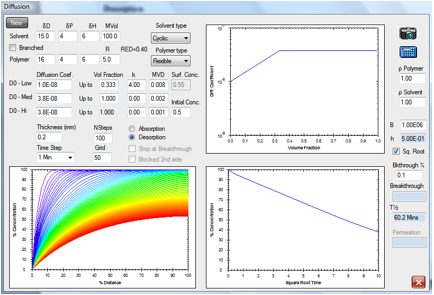
Figure 1‑5 A classic desorption curve
The same solvent is assumed to have
saturated the block of polymer and is now allowed to desorb via the left-hand
surface (the right-hand being assumed to be blocked). The coloured curves show
the solvent distribution with time, the red curve being the distribution after
100min.
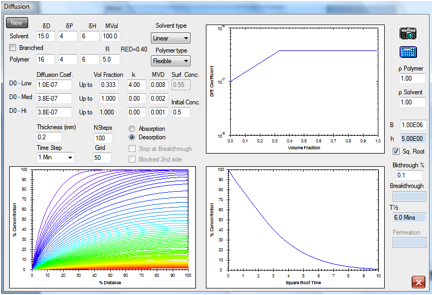
Figure 1‑6 Desorption by a smaller, faster molecule
The linear molecule, 100 molar volume,
desorbs considerably faster.
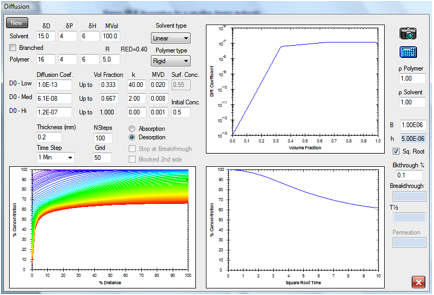
Figure 1‑7 Same molecule but desorption from a rigid polymer
This behavior resembles the formation and
drying of a polymer film from solution. Such behavior has been studied in
detail by Hansen in Hansen, C.M., A
Mathematical Description of Film Drying by Solvent Evaporation, J. Oil
Colour Chemists’ Assn., 51, No. 1, 27-43 (1968) and in the Doctoral thesis from
1967 that is available as a PDF file on www.hansen-solubility.com by
clicking on the cover page.
In a crystalline polymer, the shape is
highly skewed. Because the diffusion rates are relatively high through the
bulk, the profile is rather flat. At the edge, where concentration is very low,
the diffusion rate plummets.
“Surface
resistance”
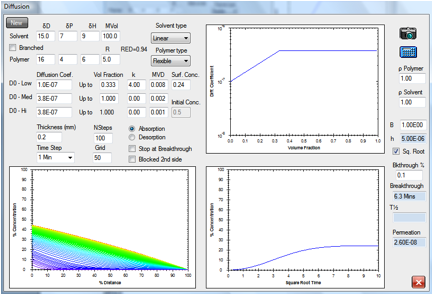
Figure 1‑8 “Surface resistance”
coming from a Mass Transfer limitation in a permeation study
In this permeation example, the “surface
resistance” comes because the “B” value (ratio of diffusivity to surface
resistance) has become significant at the entry surface. When there is a
significant surface condition at the entry surface, where there is (usually)
contact with a liquid there can also be a significant surface condition at the
exit surface, where the contact is with a gas. This possible effect at the
second surface is not considered in the present discussion. The diffusion and
surface effects are finely balanced. Solutions to the diffusion equation for
absorption, as modelled here in Figure 14-8 and in Figure 14.9, give the
so-called “s-shaped” or “sigmoidal” absorption curve. This behaviour is often
misinterpreted in the literature as being anomalous diffusion since the
absorption is not a straight line versus the square root of time, which is
called “Fickian”. The other forms of “anomalous” diffusion that are incorrectly
interpreted in the literature have the coined terminology Case II and Super
Case II. These are discussed in the following. An article in the European
Polymer Journal (Hansen, C. M., The
significance of the surface condition in solutions to the diffusion equation:
Explaining “anomalous” sigmoidal, Case II, and Super Case II absorption
behaviour, European Polymer Journal 46 (2010) 651–662) explains these
so-called anomalies with the following abstract:
“Absorption into polymers is frequently described
by the terms Fickian, sigmoidal (S-shaped), Case II, or Super Case II. This
terminology is used to describe absorption that is respectively, linear with
the square root of time, has a slight delay or S-shape with the square root of
time, is linear with linear time, or increases more rapidly than with linear
time. Solutions to the diffusion equation, Fick’s second law, that include a
potentially signficant surface condition are shown to reproduce all of these.
Sigmoidal absorption results when the surface condition is moderately
significant for either a constant diffusion coefficient or exponential
diffusion coefficients. Exponential diffusion coefficients and a lower surface
mass transfer coefficient result in Case II type behavior, with Super Case II behavior
resulting when the surface condition becomes still more significant. The
results reported here are supported by extensive experimental data with
reasonable and verifiable values for the diffusion coefficients and surface
mass transfer coefficients.”
The following figures show these effects
for absorption.
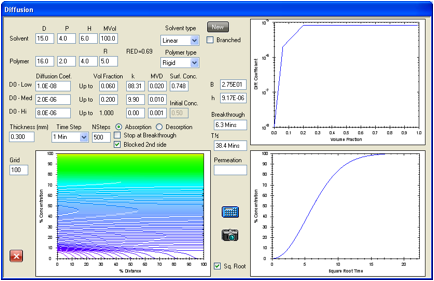
Figure 1‑9 Simulation of water absorption into untreated PVA films (Sigmoidal
Absorption).
The S-curvature in the lower right figure
matches the experimental data in [Hasimi, A., Stavropoulou K.G., Papadokostaki
M, Sanopoulou M. Transport of water in
polyvinyl alcohol films: Effect of thermal treatment and chemical crosslinking.
European Polymer Journal Vol. 44, 4098-107 (2008)] very well. The initial
curvature is very dependent on the concentration dependent diffusion
coefficients used, so improved diffusion coefficient data, especially at low
concentrations may remove any minor differences. The concentration dependent
diffusion coefficients that were used are given in the upper right corner of
the figure. The concentration profiles as a function of time confirm that there
is diffusion resistance of significance only up to concentrations near
0.1volume fraction or less. At higher concentrations diffusion within the film
is much faster than water can get to and through the exposed surface. The
experiments are primarily a measure of a mass transfer coefficient of unknown
origin (test setup, surface effects, etc.) as reported in (Hansen 2010) cited
above.
Case II absorption
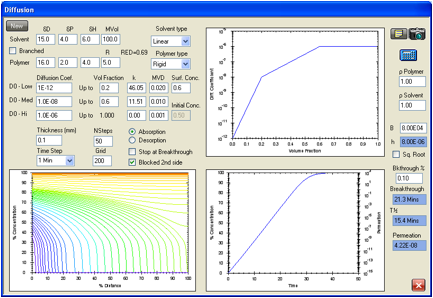
Figure 1‑10 Simulation of Case II
type absorption
The straight line
absorption curve at the lower right is typical of Case II absorption. The h
value, 8(10)-6 cm/s, is reasonable as is the diffusion coefficient
profile reported in Figure 14-1. The tail at greater than 90% absorption would
be reduced for higher h values, which could also be reasonable. This kind of
tail can be seen in the literature for the polystyrene/n-hexane system in an
often cited reference for Case II behaviour [Jacques, C.H.M., Hopfenberg, H.B.,
Stannet, V. in Permeability of Plastic
Films and Coatings. Hopfenberg, H.B. Ed. New York:Plenum Press;1974,
p.73.]. An initial curvature upward is also possible with an increased h value.
Super
Case II absorption
As we’ve stressed, there is no need to
invoke any new principles to explain exotic behaviours such as Super Case II.
To make it appear, we simply have (a) a large dependence of diffusion rate on
concentration as in Figure 14-1, which means a very low D0, and (b)
a significant surface entry resistance. Here’s a specific example:
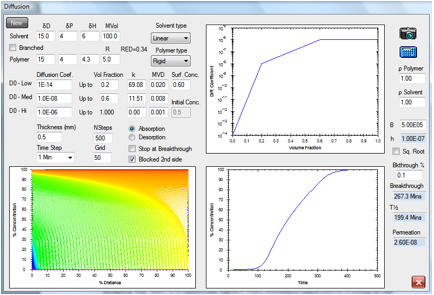
Figure 1‑11 Simulation of Super
Case II absorption
A significant surface condition coupled
with the measured concentration dependent diffusion coefficients given in
Figure 14-1 above leads to a marked increase in absorption rate well after the
absorption process has started. There is an exponential approach to the
equilibrium value at the very end of the absorption process because the driving
force for further absorption at the surface has become small. The significant
surface condition in such cases can probably be attributed to the hindered
entry resistance, perhaps like a skin, described below since the value of h is
only 1(10)-7 cm/s, but some combination with one or more additional
sources of significant surface condition resistance is also possible.
Significant
surface condition caused by hindered entry
The expected phenomena that hinder mass
transfer in absorption will also be capable of hindering mass transfer in
desorption, with the opposite sign in the mathematics, of course. These include
diffusion through a stagnant gas layer, heat transfer to or from the given
surface, wind velocity, etc. In addition to these easily recognized effects a
more subtle cause of a significant surface condition has been elucidated
recently [Nielsen, T. B. and Hansen, C. M. Significance of surface resistance in absorption
by polymers. Ind Eng Chem Res,
Vol.44, No.11, 3959-65 (2005), and also in the Handbook]. This is an entry or hindered surface passage
resistance dependent on the size and shape of the entering molecule, and, of
course, the polymer surface morphology. For smaller molecules such as
tetrahydrofuran, n-hexane, and 1,3-dioxolane there is no significant surface
condition effect of this kind for absorption into the COC polymer Topas?
6013 from Ticona. With more extensive absorption experiments it was obvious
that entry into the polymer was more difficult as the size of an absorbing
molecule increased or its structure became more complicated, such as with side
groups or cyclic entities. So-called S-shaped absorption curves, of the type
shown above, with a pseudo time-lag were observed in a number of cases,
including absorption of ethylene dichloride, diethyl ether and n-propyl amine.
Solvents containing benzene rings and more complicated structures, such as
acetophenone, phenyl acetate, 1,4-dioxane, and methyl isobutyl ketone are
completely prevented from absorbing into this same polymer, in spite of HSP
similarity to those that do absorb. This comparison indicates that they should
readily absorb into the polymer. Their absorption after prolonged liquid
exposures has been so little that it could not be detected. In such cases there
is no significant transport resistance in the external media, so it has been
postulated that there can also be a significant surface condition caused by an
entry or surface passage resistance. This type of resistance deserves much more
attention to fully understand what is happening. One can surmise however, that
for most polymers there will be smaller molecules that enter readily, and very
large molecules (such as a polymer molecule of the same kind) that will not be
able to enter at all. Between these extremes there will somewhere be a range of
molecular sizes and shapes where entry is possible but becomes retarded since
the molecules cannot rapidly find suitable sites to absorb even though they may
be adsorbed. The orientation of adsorbed molecules at such selected sites where
absorption is possible is thought to be a key element in this type of
resistance to transport. Thermal treatments can be expected to affect surface
morphology, such as in known from rapid cooling in injection molding. Because
of this, thermal treatments having an effect on surface morphology may also be
a factor in this behaviour.
Permeation
The modeller automatically calculates
permeation rates in g/cm²/s. This is of necessity an approximation
depending on how long you run your simulation – the value displayed
gradually asymptotes to the final value as the system equilibrates.
For those who prefer a plot of the integrated amount permeated that option is
available in the 3rd Edition, along with an extrapolation down the
straight part of the curve to estimate the “lag time”. Such plots are common
in, for example, the skin permeation literature. Numerous examples of
permeation in chemical protective gloves are given in Chapter 17.
Summary
With this powerful modeller you can explore
a wide range of systems, including the important “Breakthrough” type of
experiments where both kinetic and thermodynamic (HSP) factors play a role. Examples
of Fickian and what is commonly and erroneously called non-Fickian or anomalous
diffusion have been given to help guide your efforts.
The next two chapters treat diffusion in
protective gloves in more detail, showing how improved judgment of glove safety
is possible and how one can actually deduce the concentration dependent
diffusion coefficients from permeation data.
E-Book contents | HSP User's Forum
