

It was reported in the newspaper that the Omicron strain has 30 mutations that have never been seen before, and that conventional vaccines and antibodies may not work. When a person is infected with a virus, antibodies are produced, and when a new virus enters the body, the antibodies attack the virus, recognizing it as foreign.
The current RNA-vaccine, when injected, mass-produces spikes of the virus in the body. So the body is able to recognize the spike as foreign.
In antibody cocktail therapy, a mixture of several antibodies is prescribed so that one of the antibodies binds to the viral spike before it can attach to the person. As a result, they cannot bind to people.
The Omicron strain is said to have mutated this spike so that the body can no longer recognize it as a foreign body.
So, what kind of things do the spike parts bind strongly to anyway?
While searching the Internet, I found the following paper.
Identification of Dietary Molecules as Therapeutic Agents to Combat COVID-19 Using Molecular Docking Studies
It is difficult to see in a table, so please copy and paste it into Excel or something. The molecular docking energies of compounds in foods and herbs and virus spikes are calculated.
The one that binds most strongly to the spike is the tea ingredient epigallocatechin gallate (EGCG), they say.
This paper is not yet peer-reviewed, and the binding energy is a product of computer science, so it is dangerous to take it all seriously. Nevertheless, if the structure of a compound is given and its binding energy is given, there is no way to avoid analyzing it.
I will calculate the thermodynamic properties from the structural formula of Smiles using the Y-MB function in the HSPiP software, and calculate the topological properties using the RDKit in HSPiP. I am going to use RDKit to calculate the topological properties of the molecule, and then use RDKit to generate the 3D structure of the molecule. And calculate minimal cube. First, find the longest major axis of the molecule. Project each atom on the XY plane with the longest axis on the Z axis, and obtain the smallest cube with the longest one on the Y axis and the thickness on the X axis.
Drag=Rotate, Drag+Shift=Magnify, Drag+Command key or Alt key=Move.
Then, create QSAR equations using HSP, cubes, and RDKit’s Kappa1-3.
Use the nonlinear analysis tool, MIRAI.
I’ll post the data I used for those who want to see it for themselves. The last three compounds will be used for prediction.
There are 17 explanatory variables for 18 data points.
If we are going to analyze the data using the usual multiple regression method, let’s look at the results and see how many predictive values the last compound will have.
Using the method to minimize the absolute error in MIRAI, the result was as follows.
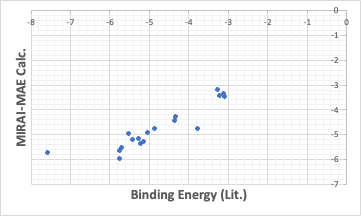
There are a few that are way off, but the others are well on the straight line, and when MAE is specified, the QSAR formula is such that the ones that are off are even further off.
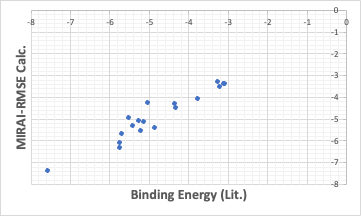
If we use an algorithm that minimizes the squared error in MIRAI, we get a QSAR formula that places everything around a straight line as much as possible.
The binding energy in these cases is stronger for smaller values. From that point of view, MIRAI-RMSE may be a better choice.
However, if there is nonlinearity, a small number of data, and a large number of explanatory variables, I would not limit the possibilities, but look at the results of several different equations and make a comprehensive judgment.
Anyway, once we have these formulas, we can calculate tens of thousands of compounds in the database and extract Kappa1-3 for HSP, cube, and RDKit.
Then I just sort and take out the most effective ones, so it all takes 30 minutes work.
The assumption is that the docking calculations are reasonably correct.
However, let’s consider whether it is better to perform docking calculations for tens of thousands of compounds to find candidates, or to do it all in one hour on a Mac mini.
It would be a much richer life if I could use my spare time for other meaningful things.
This is the kind of DX that can only be done by pirika, who seeks the richness of chemistry in digital technology.