
MIRAIはMultiple Index Regression for AIの略だ。
複数の指数関数を使った回帰式で、AI用のモデル式を作成するのに適している。
起きている事ができれば、とても役に立つことが実証されてきた。
化粧品、塗料、インクなどでは、配合処方を決めなくてはならない。しかし、使う成分はものすごく多い。
大きく分けて、例えばポリマー、溶媒、無機物、界面活性剤、添加剤などだ。
例えば、界面活性剤は何百種類とある。
他の成分を一定に保って、界面活性剤10種類を変えながら実験を10回行って、安定性、光沢、使用感などの評価を行う。
次にはその中から良さそうなものを2つ選んで使う量を変えて実験して評価する。
そして、溶媒を変える。これも何百とある。ポリマーを変える。添加剤を変える。
そうした結果が特許などに記載されている。
Materials Informatics(MI)を使って、こうした処方設計を効率化する事を考えてみよう。
まず、ビッグ・データを用意する。
ポリマー、溶媒、無機物、界面活性剤、添加剤が10種類ずつあれば、10*10*10*10*10の組み合わせがあるので、100,000の組み合わせがある。
使う量を10水準(例えばポリマーを20%から40%まで2%刻みで評価)変えれば、100,000の組み合わせ1つに対して100,000水準があるので、1010の結果がありうるので、とてもビッグだ。
現実のところはどうかというと、成分は最大で500、実験数は最大で1000、評価項目は最大で50種類ぐらいらしい。
しかも、一つの実験で使っている成分はせいぜい20種類ぐらいで、残りの480成分は値がゼロになる。つまり500*1000のデータのうち4%ぐらいしか値のない、スカスカのテーブルになっている。
やりたい事は単純だ。
評価値1=f1(X1, ,,,, X500)という関数fを各評価ごとに求める。
誤差のトータル = Σ|実験値の評価1 - f1(X1, ,,,, X500)| 実験の個数分だけ足す
関数f1は誤差のトータルが最小になるように定める。(誤差は絶対値か2乗誤差)
どの成分が評価をどれだけ上げ下げするか?
これが定量的に分かれば、コンピュータを使って逆解析すれば良い。
つまり、f1(X1, ,,,, X500)が求まれば良い。
次元の縮退
特許などを解析しようとすると、実験数よりも成分数の方が多いことも多い。
連立方程式の解法を考えても、式の数は変数の数より多い必要がある。
そこで、PCA(主成分分析)やPLSを使って次元を縮退させることをやるのが普通だろう。しかし、界面活性剤が実験で1回しか使われていないなら、種類の違う界面活性剤の実験結果は直交(内積がゼロ)してしまうので次元は減らせない。
解析ツールMIRAI
MIRAIに関しては次のような記事を書いているので、詳しいことはそちらで読んで欲しい。
ブログ:MIRAIによるMI(マテリアル・インテグレーション)研究
ブログ:塗料、化粧品、インクなどの配合処方設計のAI化
固定ページ:配合処方設計トップページ
このブログで触れたいのは、全く別の事だ
MIRAIを使った成果
こと、配合処方を最適化するのにMIRAIは素晴らしい成果をあげることが利用企業で明らかになってきた。
成分数400超の実験を解析し、モデル式を作成し、しかも、誤差が大きいものを調べたら皆入力ミスであったことがわかった。
評価項目も30以上あり、並行して1台のマシンにjobを6個(Core数まで)流す事も始まった。さらに利用するコンピュータを増やす計画もある。
(山本に報酬を6人分くれないかな。。。)
どの成分が、どの評価の時どういう係数になるかを比べると非常に早く成果に繋げることができる。人間がカンと経験で培ったものを、人が納得できる形で定量化できる。
在宅勤務になったら、帰る前にjobを放り込んで計算させておくことができる。
Pirikaのツールを使って化学をもっと豊かにDXを地でいく成果だ。
DXを行って生産性向上というのはこういう事を言うのだろう。
問題発生
ところが、問題が発生。MIRAIの問題点は、計算に時間がかかる事だ。そしてブラウザーの上で動くWebアプリとして提供していることだ。
そして、利用している企業は、セキュリティー対策のため、誰も操作していない場合には自動的にロックがかかるようになっている。そしてそれを解除するのは禁止ということだ。
ソフトを開いて、キーの上に重しを乗せて操作され続けているように偽装する、など対処法が考えられた。
ちょっとあんまりなので、ソフトが起き続けるように改造を行なった。
前振りが長すぎ
WakeLock実装
大した事をしたわけでは無く、JavascriptのWebLockを実装しただけだ。
これは、webアプリで例えば地図見ながら歩いていたら、操作しないので1分で画面がオフになった。なんて事がないように、擬似的に操作を与え続ける機能のようだ。
(MacのSafariには実装されていないようなので、Chromeを使う)
使い方は簡単で、bodyタグにwake lockを発動するボタンを配置する。
<div>
<button id="btnStart" class="button">Screen Wake Lock をスタートする </button>
</div>
<div class="demo">
<p>
Screen Wake Lock status:
<span id="status"></span>
</p>
</div>
そして,javascriptで次のように記載する。
<script type="text/javascript">
if ('wakeLock' in navigator) {
let btnStart = document.querySelector('#btnStart');
let status = document.querySelector('#status');
let wakeLock = null;
//---------------------------------------------
// screen wake lock をリクエストするための関数
//---------------------------------------------
const requestWakeLock = async () => {
// ブラウザは Screen Wake Lock を拒否することがあるので、
// try...catch を使い拒否された場合の処理も記述する
try {
// screen wake lock をリクエストする
wakeLock = await navigator.wakeLock.request('screen');
// Screen Wake Lock がリリースされたときの処理
wakeLock.addEventListener('release', () => {
status.innerHTML = '<span style="color:red;font-weight:bold;">released</span>';
});
status.innerHTML = '<span style="color:green;font-weight:bold;">active</span>';
} catch (err) {
console.error(`${err.name}, ${err.message}`);
}
};
//--------------------------------------
// Start ボタンをクリックしたときの処理
//--------------------------------------
btnStart.addEventListener('click', async (event) => {
// screen wake lock をリクエストする
await requestWakeLock();
});
}
</script>
すると、下図のようにボタンが出てくるので、クリックするとステイタスがactiveになる。これで、一晩でも、週末全部でも働き続ける。
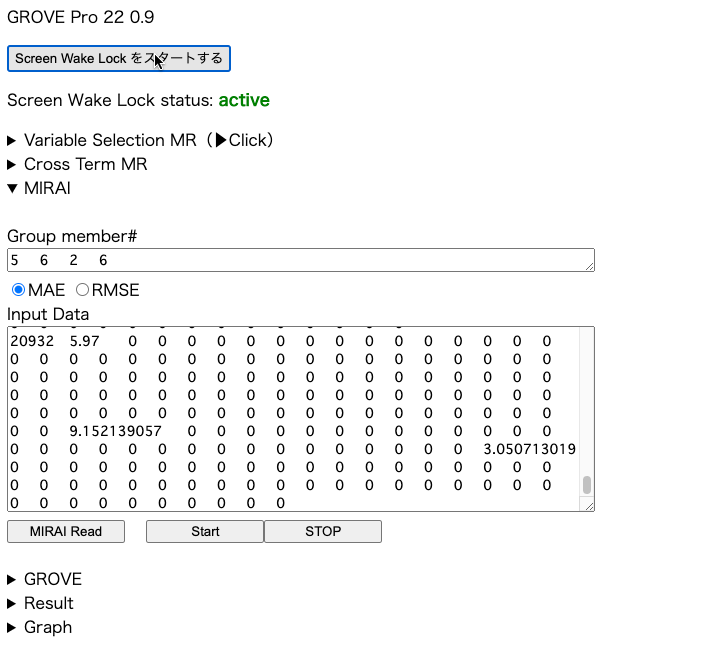
このGROVE proはpirika研究会に参加している企業、アカデミックが利用可能になっている。