
2025.07.5
pirika.comで化学
>チャピエモン-3rd Pirika Origin (C-3PO)
> ハンセン溶解度パラメータ (HSP)
>HSPiP(実践ハンセン溶解度パラメータ)ソフトウエアー
> HSPiPの購入方法
> HSPiPを用いた解析例
>基礎 >応用 >ポリマー >医薬品など >環境・溶媒設計 >分析
>その他 >DIY/YMB >アバターチュートリアル >次世代に向けて
>次世代HSP2技術
> 化学全般
>Pirika Pro ツール群
ブログ
業務案内
お問い合わせ
注意:HSPiPの機能ではありません
分子軌道計算
私は1990-1991年にCALTECHのGoddard教授のもとに留学した。
もともとは高分子の合成屋だったので、とんでもなく大変な留学だった。
取り敢えず、分子軌道計算や分子力学(MM)計算はこなせる。
1995-1999年にはつくばに出向して代替フロンの研究をしていた。その際にはフロンの分解をMOを使って計算していた。
MO計算、特に遷移状態計算はとにかく時間がかかる。そこでジョブを放り込んでは後はニューラルネットワーク法のプログラムを組んだりして情報化学系にのめり込んでいた。
個人的には、MOPAC程度の半経験的分子軌道法の方が計算が早くて好きだ。精度が低いとか色々言う人はいるが、どんなに厳密なMO計算したところで、沸点すら予測できない。それなら、立っているものは親でも使え。さまざまな情報源の一つとしてMO計算を捉える。そして代替フロンの設計に活かした。
今の時代では、コンピュータの速度はAb Initio計算であっても十分早い。
広い原子にも対応しているので、ちゃんとした分子軌道計算と合わせるのは良い選択だろう。
Y-MBPro物性推算
もともとは、代替フロンの物性を推算するため、”The Properties of Gases and Liquids“を紐解いていた。だんだん、フロンとは関係ない独自の世界に入り込み、1999年にはこのpirika.comを作り、Web top Computingのサイトとして認知されるようになった。このpirikaではMO計算(デジタル分子模型)も有名である。
橋渡し
pirikaの物性推算のページでは最初はお絵かきソフトを自分で作り、描かれた分子を解析して物性推算を行っていた。更にその前は原子団の数を自分で入力する方式だった。すると、分子をこちらの意図しない所で切断されることがある。
ちなみに、YMBでは大きな原子団を先に定義していく。ter-Butyl (CH3)C(CH3)(CH3)をCH3を3個と>C<を1つとはカウントしない。
お絵かきから、独自のMolテーブルを作成し、テーブルを解析して原子団なり原子結合テーブルを作り物性値と結びつけた。
その後、分子の構造としてはSMILESの構造式を用いることにした。一分子1行で扱えるのは嬉しい。
困るのがMO計算だ。1次元のSMILESから3次元構造を作らなくてはならない。
簡単なMM計算でCleanすることも試みたが余りうまくいかなかった。
現在はRDKitがSMILES構造式から分子の3次元構造を吐き出してくれる。RDKitの分子構造はいわゆるmol2フォーマットだ。これはOpen Babelを使えば簡単にガウシアンなどの入力フォーマットに変換できる。
つまり、分子をSMILESの構造式で蓄えているような情報系の化学者には既に橋渡しはできている。
分子軌道計算屋からの橋渡し
最近、DXやらMIやらで、分子軌道計算屋さんが情報系の仕事をすることが世間でも増えてきている。これまでに計算した膨大な分子のDBを持っているかもしれない。そうして得られた最適化した3次元構造は、とても重要なものだろう。座標データを捨て去ってSMILESだけDBにすればいいというわけにはいかない。
高価な分子設計支援システムを導入すればどうにかなるのかもしれない。
(ぼくは、この支援ってだっきらいだ)
正確な構造データを残しつつ、MIをすすめる
分子軌道計算屋さんの顔を立てつつ、情報系の仕事の加速化する。
DB化もしやすいように。
ということで、JSONで1行の文字列化したsdfをYMBで扱えるようにしてみた。
sdfフォーマット中の分子構造とは?

Molファイルとの違いは私にはよく分かっていない。ただ、MolとかMol2とかはとても混乱しているように思える。座標が画面上の座標であることも多い。sdfと言ったときには、座標は分子軌道で用いられる座標のような気がする。
フォートランの名残か、原子数、結合数の位置は厳密で、結合数が99を超えるとどこまでが原子数でどこから結合なのかはわからなくなる。
通常は各分子について一つのsdfファイルが存在する。
sdfファイルにファイル名を入れる

S-Projectから500化合物ぐらいのsdfファイルが送られてきた。ファイルの中にはファイル名が無かったので、1行目にファイル名を付け加える。最初は手作業で始めたが余りに面倒なのでChatGPTにやり方を聞いた。瞬間で500化合物の処理が終わる。
sdfファイルの連結
>cat *.sdf>MyAll.sdf
Unixのcatコマンドを使えば全てのファイルを連結できる。
分子の区切り記号は$$$$になる。
連結されたsdfsのファイルの解析
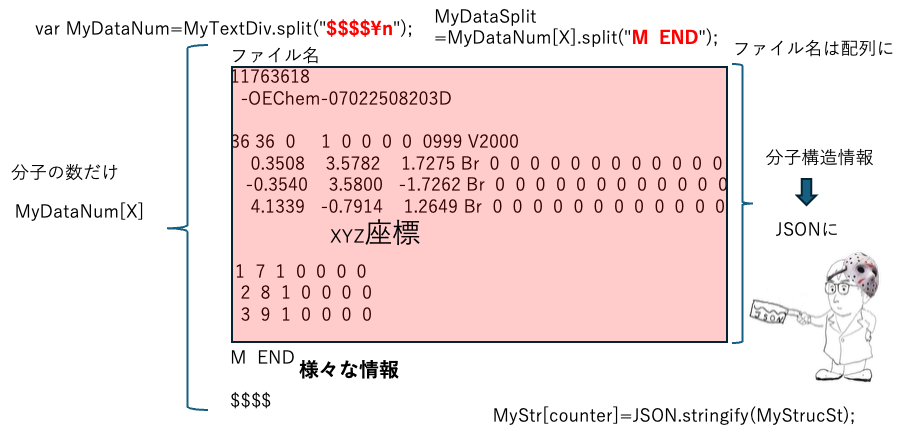
ファイルを$$$$で区切り、さらにM ENDで区切り分子構造のみの部分をJSON文字列にして配列に保存する。結果をExcelに貼り付ける。
1つのセルに分子の3次元構造データ
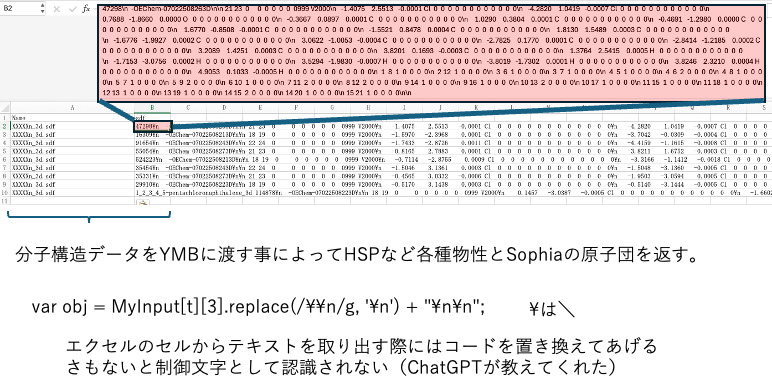
A列にはファイル名、B列にはJSONで文字列化された分子の構造データが入っている。
YMBの入力に(JSON文字列化された)sdf
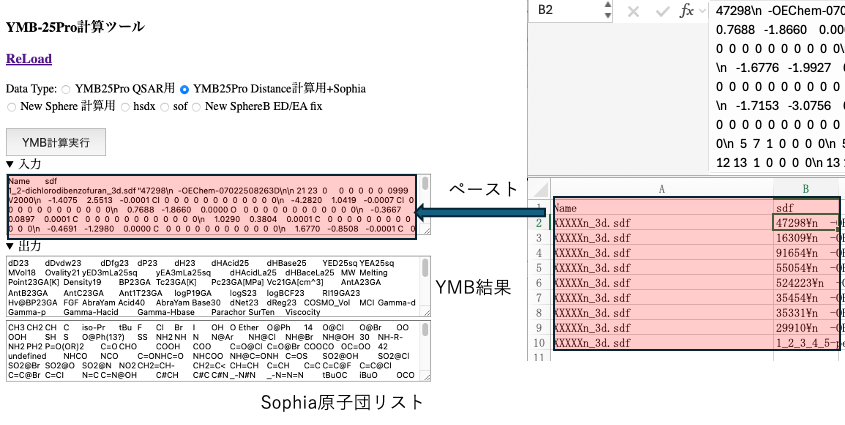
エクセルから分子データをペーストして計算を実行すると、Smilesの時と同じようにYMBの計算結果とSophiaの原子団リストを出力してくれる。
MOの計算結果とYMBProの出力
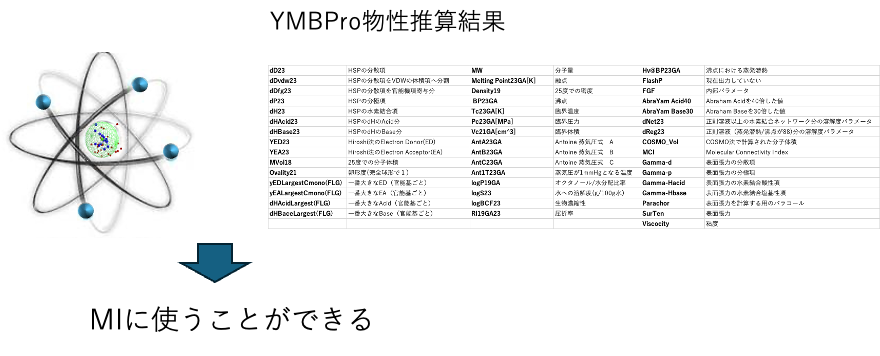
MO計算の結果とYMBの熱物性の両方を使いMIを進めることが出来る。
たとえば、毒性や薬効を予測するのに、LUMOのエネルギーと溶解性の指標を両方使うことが簡単にできるようになる。
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)
メールの件名は[pirika]で始めてください。