
2026.3.30
pirika.comで化学
>チャピエモン-3rd Pirika Origin (CPO)
> ハンセン溶解度パラメータ (HSP)
> 化学全般
> 情報化学 >物性化学 >高分子化学 >化学工学 >その他の化学 >昔のもの
>情報化学ツール >MAGICIAN養成講座 >STEAM
>Pirika ツール群
ブログ
業務案内
お問い合わせ
情報化学ツール > 原子団寄与法>
原子団寄与法の基礎
Dynamic Group Contribution > DGCの使い方
DGCの原子団拡張
DGCの応用例
[1. 概要]
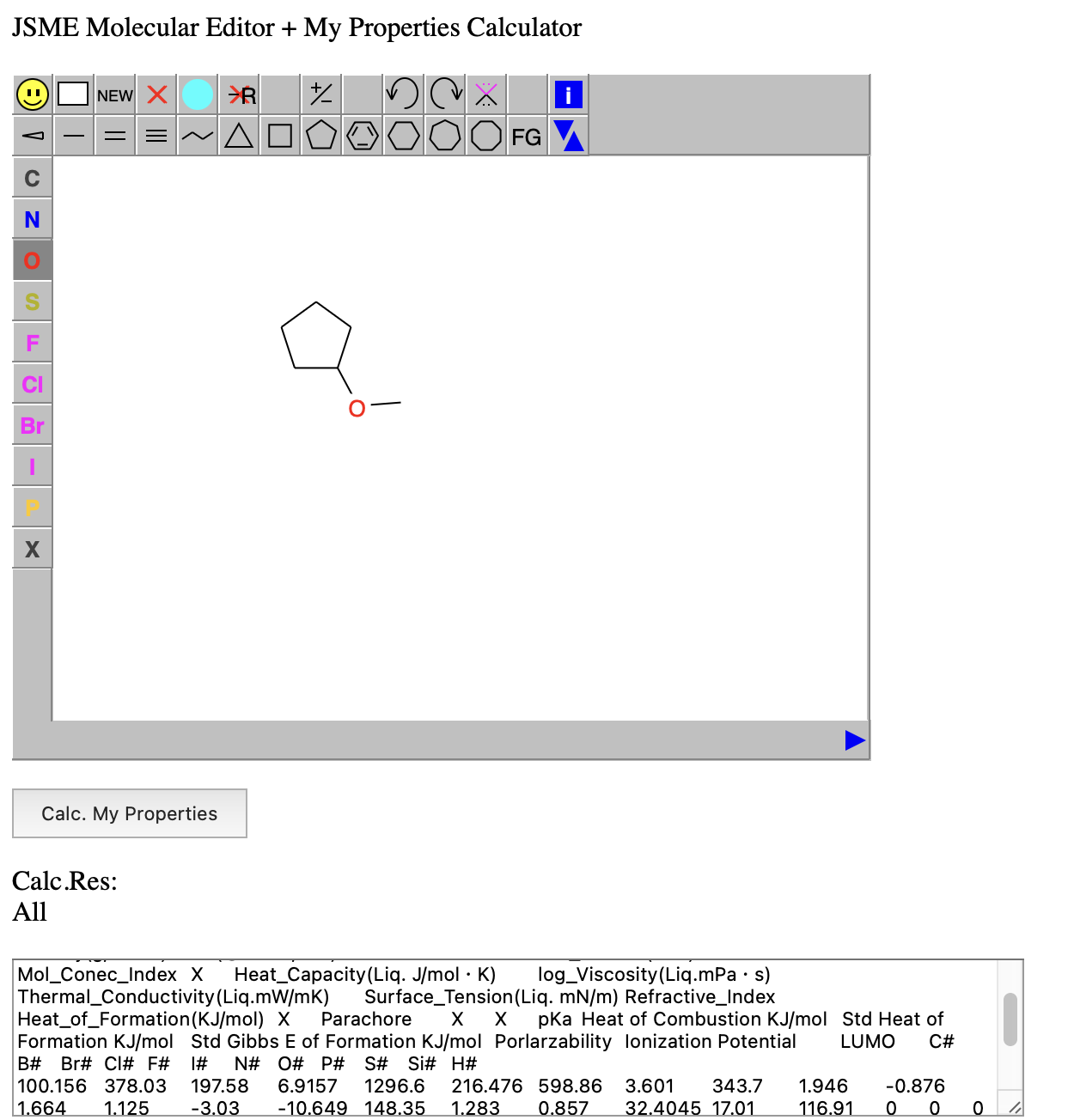
Dynamic Group Contribution(DGC)法を用いて物性推算を行うには、まずSMILESの分子構造式[*1]と物性値のペアを用意する。ここではpKaで説明する。表1に示すような909化合物のデータを用意する。このSMILESの構造式をYMB4DGC[*2]で処理すると表2に示すDGC用の結合テーブルが作成される。この結合テーブルをDGCに入力し計算を行う。結果は予測値のテーブル、実際にSMILESの分子構造から物性値を予測するプログラムを出力する。他の方法でも検討できるようにテーブルも出力する。
[2. Smilesの構造式から結合テーブルを作成する]
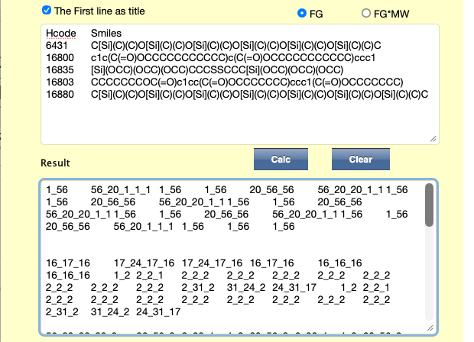
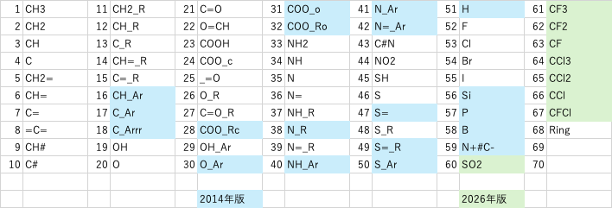
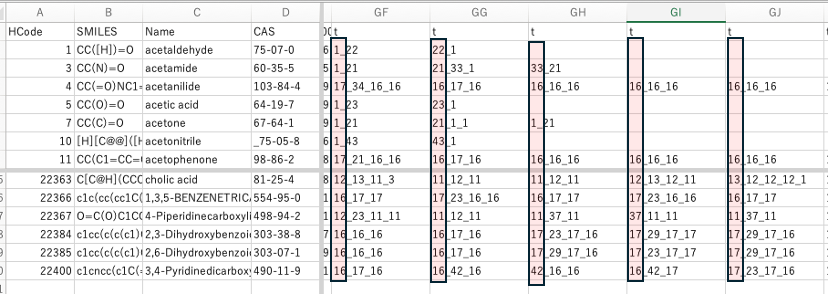
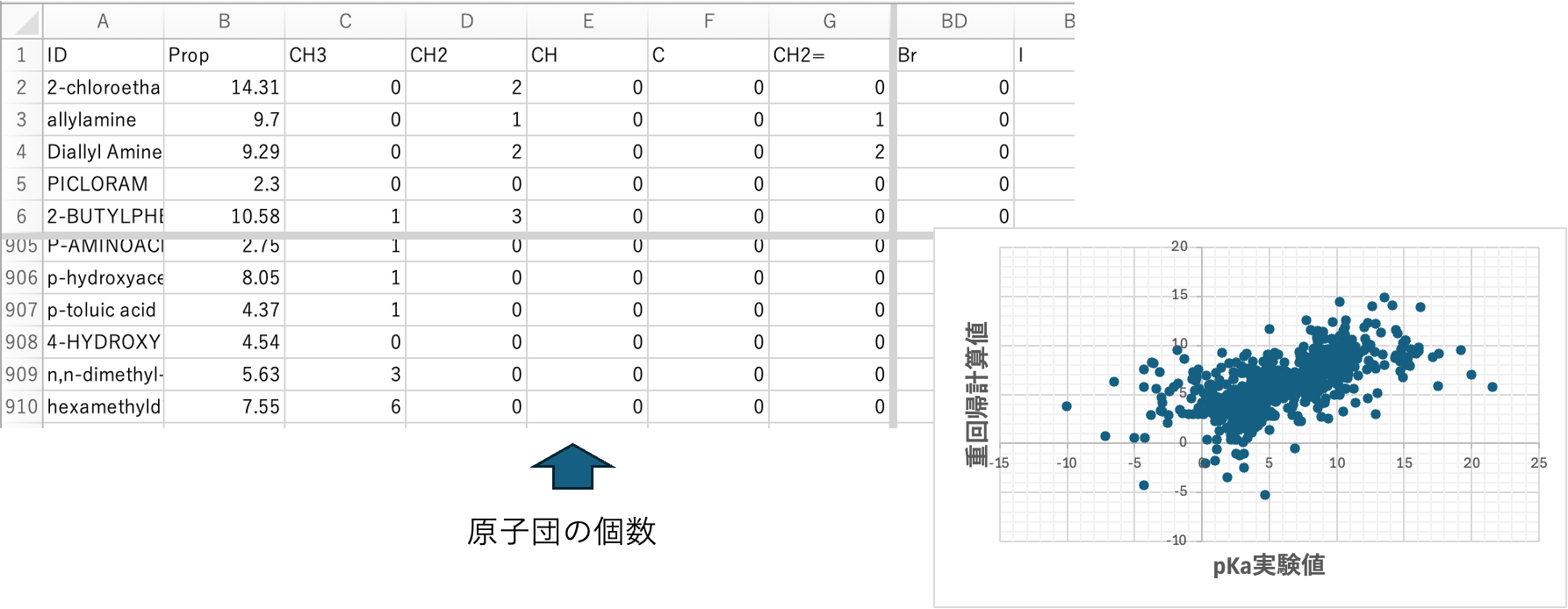
今回(2025年)pKaのデータを解析した。SMILESの構造式があれば、2026年バージョンYMB4DGC(図1)では67種類(2014年バージョンでは59種類)の原子団を使って結合テーブルを自動的に生成する。結合テーブルは次の形式で表現する。最初の数字は表3に示すメイン原子団の番号になる。メインの原子団は結合数によって(複数の)subの原子団が付加する。表4に示すように最初の数字はメインの原子団その後は_を介して(複数の)原子団が付加した結合テーブルが作成される。
[3. 結合テーブルをDGCで読み込む]
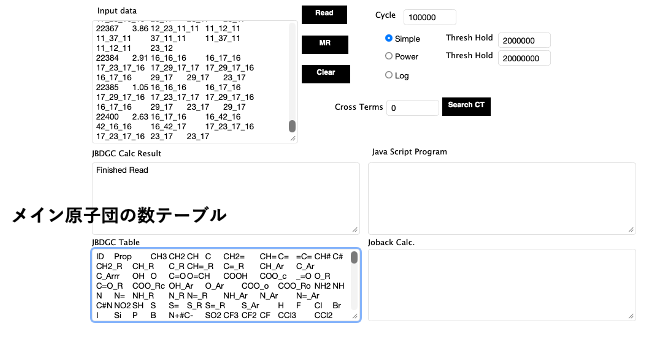
結合テーブルをDGCで読み込むと図2に示すメイン原子団だけのテーブルが作成される。この67種類の原子団から原子団寄与法で物性推算することができる。その精度はJOBACK法[*3]程度になると予想される。メイン原子団の数だけから重回帰法で物性値を予測すると図3になる。精度は高いとは言えない。例えばカルボキシル基にCH3が付加する場合とCF3が付加する場合でpKaの値が大きく異なる。メイン原子団だけに切断してしまうと、COOHの係数は-2.408と1つに決まってしまう。この係数は様々な種類のカルボン酸の平均値になっている。
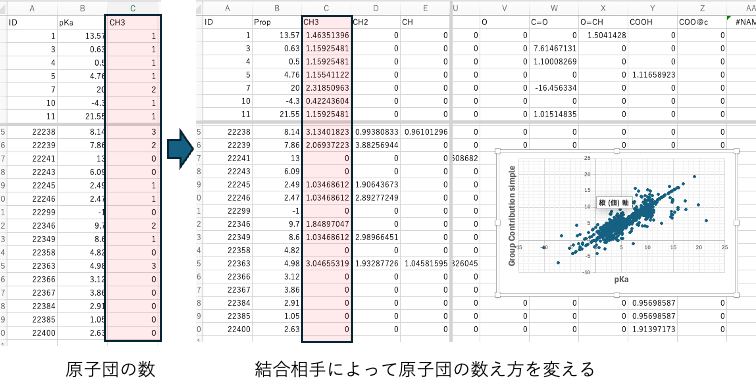
[4. 相手によって原子団個数の数え方を変える]
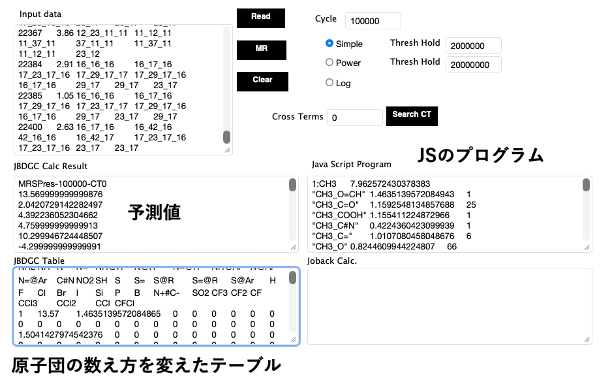
DGCを使って結合相手によってメインの原子団の個数の数え方を変える。収束計算になるので計算時間は長くなる。気長に待つ。計算が終了すると図4に示す結果が表示される。結合相手によって原子団の数え方を変えたテーブルが図4の左下に表示される。図5に示すように、元は原子団の個数そのもののテーブルから結合相手によって個数が変更されたテーブルになる。図4の予測値と元の物性値をプロットすると図5のグラフのようになる。図3のグラフと比べ大きく改良されていることがわかる。特に小さな分子、例えばCH3OH(pKa実験値15.3)は, DGC予測値は15.3になる。変換テーブル中のCH3_OH, OH_CH3は分子そのものになるので予測値は実験値そのものになる。そこで小さな分子は実験値と予測値がグラフの対角項に並ぶので見かけ上精度が高くなる。
[5. 非線形変換オプション]
原子団の数え方を変えた後、非線形変換(Power関数、Log関数)する事も可能である。このオプションは効果がないことも多い。
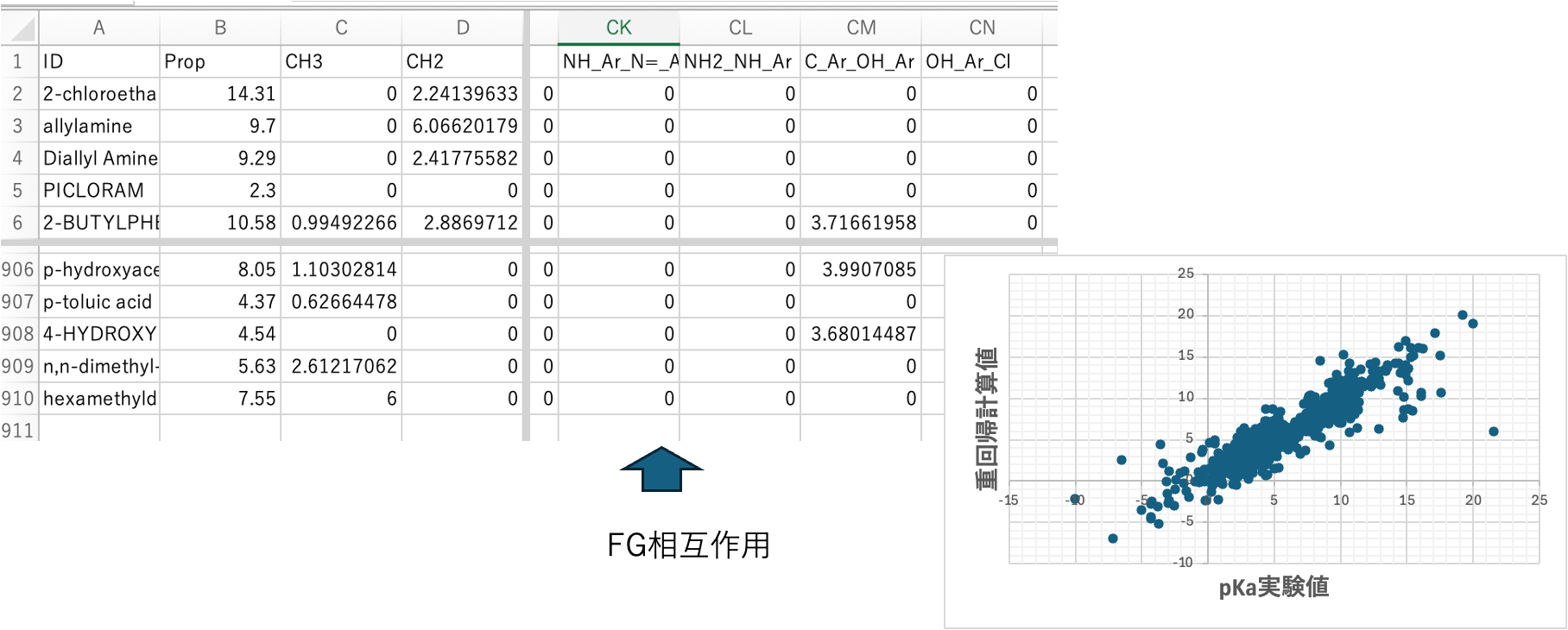
[6. メイン原子団同志の相互作用]
R-CH(NH2)COOHのアミノ酸とNH2-CH2CH2-COOHを考えてみる。CHの数え方CH(NH2, COOH)で一つの炭素にアミンとカルボン酸が付くことの補正は行われる。NH2-CHとCOOH-CHの補正は行われる。でも一つの分子(NH2-CH2CH2-COOH)の離れた位置にNH2とCOOHの両方を持った時にどうなるかは、原子団の数え方では補正できない。そうした相互作用を幾つ仮定したら良いかは一概に言えない。クロスターム項を20導入すると指定してDGCソフトに20ペアを探索させる。67原子団から2つのペアを選ぶには67*66通りある。そのペアから20ペアを選ぶ。4422C20の中から一番pKa予測誤差が小さくなる20ペアを選ぶ。グローバルミニマムを探索するのは難しいので遺伝的アルゴリズム法[*4]を用いて探索する。ここではCOOH_OH@Ar, COOH_COOH, CH2_CH2, C@Ar_COOH, COOH_NO2, CH2_OH@Ar, OH@Ar_OH@Ar, NH2_NH2, CH@R_NH@R, C@Arrr_OH@Ar, CH@R_NH, C=_NH2, C=O_C=O, CH@Ar_O, O_O, CH2@R_F, C@Ar_NH, O=CH_O=CH, N=@Ar_P, N=@R_Clの20ペアが選ばれた。結果は図6に示す。図5と比べると大きく外れるものは減るが、実験値と予測値が一致していた低分子では予測値が外れてしまう。クロスターム重回帰[*5]自体はpirikaの別ソフトとして提供している。
[7. DGCの計算機]
DGC計算を行えば、図4に示すように結果はプログラムとして出力される。それをpKa.jsとセーブする。図7に示すようにプログラムを計算機に組み込めば、分子の絵を描けばpKaをすぐに予測することが可能になる。
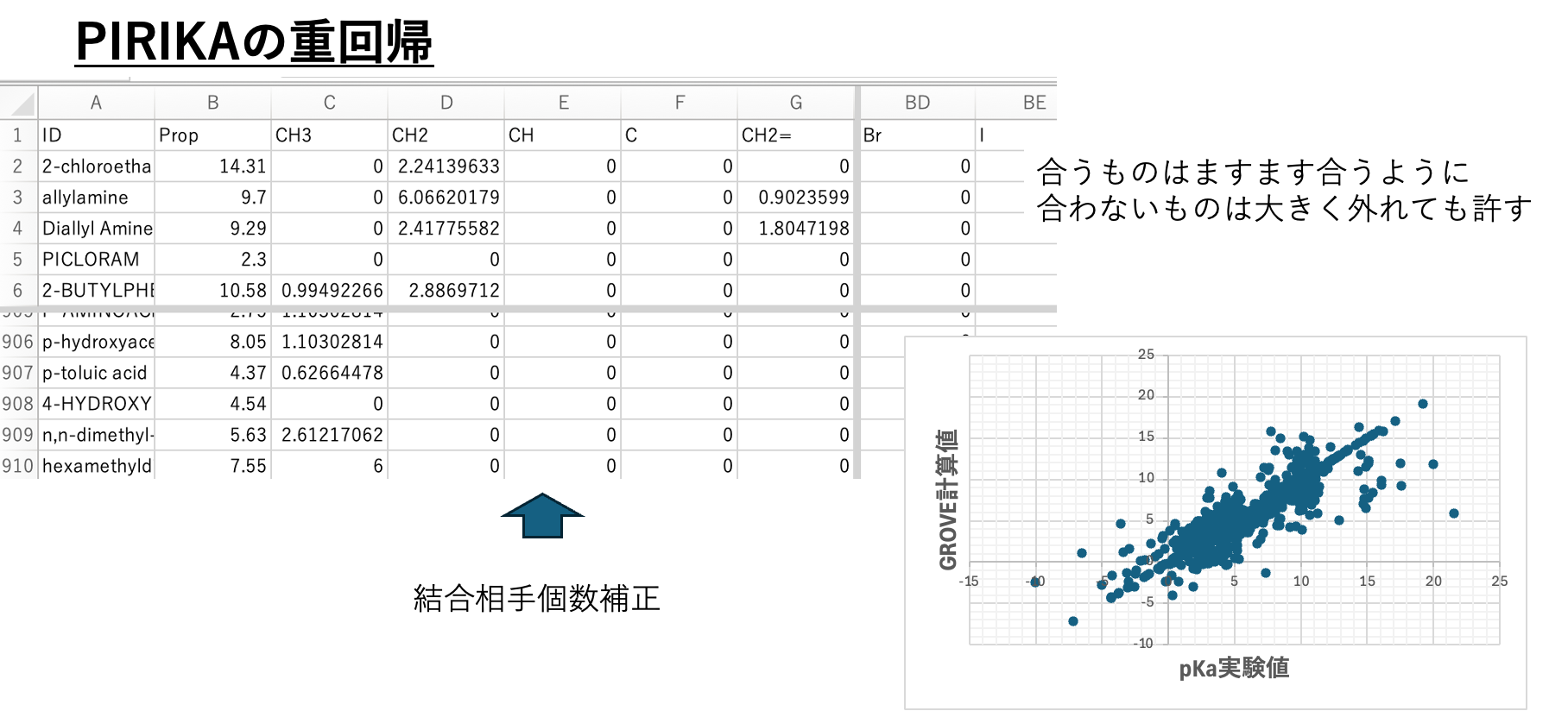
[8. 重回帰の係数を最適化(GROVE、LASSDGE)]
通常の重回帰は二乗誤差を一番小さくするように動作する。実験データが正しくない場合、誤差がとても大きくなる。その大きな誤差を小さくするように重回帰は動作する。
そこで実験データに誤差が含まれる場合には、通常の解析では正しい答えにならない。pirikaのGROVE法[*6]では合うものはますます合うように、合わないものは大きく外れても許すので結果の解釈が容易になる。さらに係数の最適化まで行うLASSDGE[*7]を使うこともできる。結果を図の8に示す。
[9. 図表]
| HCode | SMILES | Name | CAS | EPIDataBase::PKA |
| 1 | CC([H])=O | acetaldehyde | 75-07-0 | 13.57 |
| 3 | CC(N)=O | acetamide | 60-35-5 | 0.63 |
| 4 | CC(=O)NC1=CC=CC=C1 | acetanilide | 103-84-4 | 0.5 |
| 5 | CC(O)=O | acetic acid | 64-19-7 | 4.76 |
| 7 | CC(C)=O | acetone | 67-64-1 | 20 |
| 10 | [H][C@@]([H])([H])C#N | acetonitrile | _75-05-8 | -4.3 |
| 11 | CC(C1=CC=CC=C1)=O | acetophenone | 98-86-2 | 21.55 |
| HCode | pKa | BT | BT | BT |
| 1 | 13.57 | 1_22 | 22_1 | |
| 3 | 0.63 | 1_21 | 21_33_1 | 33_21 |
| 4 | 0.5 | 17_34_16_16 | 16_17_16 | 16_16_16 |
| 5 | 4.76 | 1_23 | 23_1 | |
| 7 | 20 | 1_21 | 21_1_1 | 1_21 |
| 10 | -4.3 | 1_43 | 43_1 | |
| 11 | 21.55 | 17_21_16_16 | 16_17_16 | 16_16_16 |
[10. Pirika.comへのリンク]
*1: SMILESの分子構造式
*2: DGC結合テーブル作成用のYMB4DGC
*3: Joback法
*4: 遺伝的アルゴリズム法
*5: クロスターム重回帰
*6: GROVE法
*7: LASSDGE法
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)
メールの件名は[pirika]で始めてください。