
2026.3.25
pirika.comで化学
>チャピエモン-3rd Pirika Origin (CPO)
> ハンセン溶解度パラメータ (HSP)
> 化学全般
> 情報化学 >物性化学 >高分子化学 >化学工学 >その他の化学 >昔のもの
>情報化学ツール >MAGICIAN養成講座 >STEAM
>Pirika ツール群
ブログ
業務案内
お問い合わせ
情報化学ツール > 原子団寄与法 >
原子団寄与法の基礎
Dynamic Group Contribution
DGCの原子団拡張
DGCの応用例
AIよ。Dynamic Group Contribution (DGC)をPirika proに実装する準備ができた(ブログからの転記)
AIよ。pKaの推算方法を人間に教えてやってくれ。(ブログからの転記)
[1. 概要]
原子団寄与法は原子団を先に定義する必要がある。
物性推算の精度を上げるのに大きな原子団を導入するのが効果的だ。しかし原子団を増やすにはデータセットも増やさなくてはならない。DGC法[*1]では原子団の結合の違いを見るのでメインの原子団の定義数を増やす必要はない。例えばアミド基は定義していない。しかし、C=OとNH2, NH, Nが定義されていれば、C(=O)NH2, C(=O)NH, C(=O)Nが定義されているのと同じになる。NH2, NH, Nが芳香族についた場合にも結合の相手が異なるだけなので定義し直す必要もない。データセットによってダイナミックに定義が構築される。ただし、原子団の種類によっては精度が出ない事がある。その時には新たに原子団を追加しなくてはならない。必要に応じて原子団の数を増減しなくてはならないので、手間のかからない方法にしなければならない。
[2. SMILESの構造式の解析]
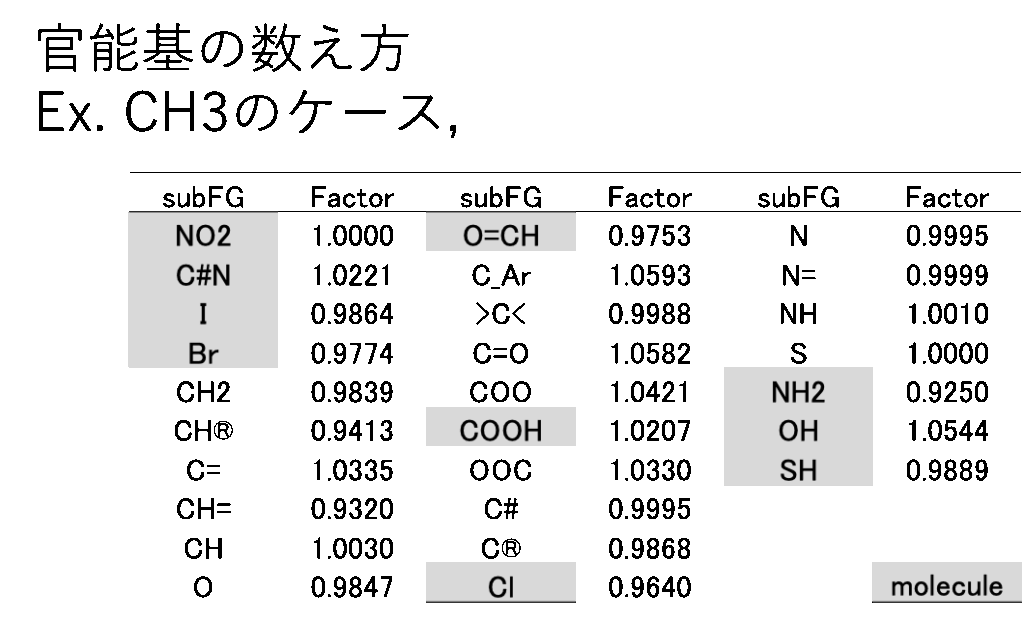
Pirika Proに搭載されているYMB[*2]はSMILES[*3]の構造式の大きな原子団から認識させていく。例えばt-ブチル、2-ブチル、2-ブチル、i-ブチルを認識させたいとする。4級炭素を探してそれに接続するものがメチル基3つだったらt-ブチルがある。3級炭素にメチルが2つ、CH2が1つ接続していたらi-ブチルというように認識させていく。NOを両方持つものとしては NO2、SOを両方持つものはSO2, S=Oの順に認識させていく。芳香族の認識は少し難しい。認識した原子団がどんな原子団と結合しているかの情報を元に結合テーブルを作る。「The properties of Gases and Liquids 3rd 」という書籍には400化合物のデータがある。そのデータ中でCH3がどんな原子団と結合しているかをリストアップすると表1のようになる。CH3にケトン(C=0)が付いた時にCH3は1.0582個あると考える。CH3にアミン(NH2)がつくと分子そのものになる。その時のCH3の数え方は0.9250個あると考える。メインの原子団にどんな原子団が結合するかは、データセットに依存する。係数が求まっていないペアを予測するには係数は1.0を使う。最悪JOBACK法[*4]程度の精度が確保される。
[3. DGCで精度の出ないケース]
例えばCH3-CF3という分子を考える。CF3という原子団は(以前のバージョンでは)定義されていない。そこで、CH3は4級炭素と結合していると数えるしかない。>C<の係数は0.9988になる。>C<に結合するCH3は0.9988と数える。CF3でもtBuでも>C<に変わりはないのでCH3の数え方は同じになる。それで問題がでる物性値もある。
CF3COOHのような化合物のpKaで問題が出ていた。
COOHが>C<に接続したと考えるとCOOHがCF3によって電子が引っ張られるという効果は導入されない(おまけの図で電荷平衡法[*5]でのチャージ参照)。>C<_F_F_Fの時に>C<の数え方が大きく変わるかが問題になる。
もともと、pKaに影響を与えるのは活性水素を持つような原子団だけだ。それ以外はほとんど影響を与えない。たとえば、CH3CH2CH2CH3とかCF3CF2CF3はpKaに何の影響も与えない。そこで>C<_F_F_Fがあっても平均をとると>C<の数え方はあまり大きくは変わらない。そこでCF3は定義する必要がある。N, P, Sのように価数が変わる化合物も厄介なので、メインの原子団として定義してしまいたい。
[4. 原子団の定義拡張]
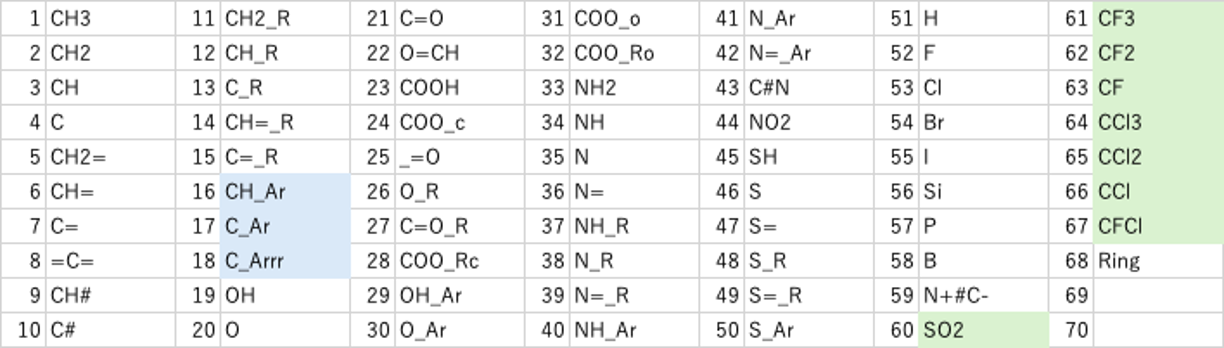
元々、YMBでは172種類の原子団[*6]を定義していた。その原子団をSMILESの構造式から認識させるプログラムは既にある。少し調整すればDGC用に新しいテーブルを作るのは簡単だ。表2に示すように、今回SO2, CF3, CF2, CF, CCl3, CCl2, CCl, CFClの原子団を増やし67原子団に拡張子た。(ringは除くことにした。)そして、DGCを実際に使ってみる。合わないものをなぜ合わないのか考える。どう改良したら合うようになるか試す。
- SMILESの構造式から原子団を数え上げてテーブル化する。
- 原子団同士の結合情報をもとにDGCで推算式を構築する。
- 推算式を用いて任意の化合物の物性を推算する。
- 合わないものを何故合わないか考える
これを繰り返す。
原子団の定義の増減によってプログラムの修正が必要ないようにプログラムの修正を行った。ユーザーが持つデータをDGCで推算式を作る。原子団の拡張が伴う事がある。
たとえばN+やCl-のようなDES用の拡張が簡単にできるようにする。
[5. 図表]
[6. pirika.comのリンク]
*1: Dynamic Group Contribution
*2: YMB Yamamoto Molecular Break 山本の作った分子の分割アルゴリズム
*3: Smiles線形表記法について (HSPiP Docs) Smilesの分子構造式 (MOOC)
*4: Joback法
*5: 各原子上の電荷を計算 電荷平衡法
*6: 官能基のリスト (TCPE), HSPiPで使える官能基一覧 (HSP/Docs)
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)
メールの件名は[pirika]で始めてください。
[7.おまけ]
これまでの原子団寄与法では、例えばトリフルオロ酢酸(CF3COOH)と酢酸(CH3COOH)をCF3+COOH、CH3+COOHという原子団に分ける。カルボン酸(COOH)は酸なのでpKaを小さくする原子団になる。
ところが、トリフルオロメチル(CF3)はいろいろな所で使われていて、例えば、CF3-CF3という化合物ではpKaに何もしない。メチル(CH3)も同様だ。その平均値で考えてしまうと2つの化合物のpKa計算値に差は出ない。
piriakで開発したDGC(Dynamic Group Contribution)を使うとpKaに関してはCF3COOH(予測値:0.724 実験値:-0.25)
CH3COOH(予測値:4.71 実験値:4.76)
と推算できる。
結合している相手によって、原子団の数え方をダイナミックに変化させているのがミソだ。難しいMO計算はいらない。分子を描くだけだ。
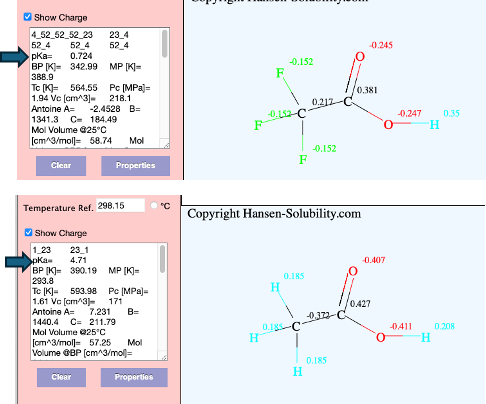
ここではまだCF3は定義していない。