
2025.7.9
pirika.comで化学
>チャピエモン-3rd Pirika Origin (CPO)
> ハンセン溶解度パラメータ (HSP)
> 化学全般
> 情報化学 >物性化学 >高分子化学 >化学工学 >その他の化学 >昔のもの
>情報化学ツール >MAGICIAN養成講座 >STEAM
>Pirika ツール群
ブログ
業務案内
お問い合わせ
情報化学ツール > 原子団寄与法>
原子団寄与法の基礎
Dynamic Group Contribution
DGCの使い方
DGC結合テーブル作成用のYMB4DGC
DGCの原子団拡張
DGCの応用例
[1. 概要]
原子団寄与法(Group Contribution method)の問題点は切断することによる情報の欠落だ。CH3CH2CF2CF3とCH3CF2CH2CF3で原子団の数は同じになってしまう。物性値によってはこの違いはとても大きい。より大きな原子団を定義すれば解決できるが原子団を増やすと必要なデータ数も多くなる。原子団を増やさず結合情報を保つ、Joback法[*1]の改良法をDr. Jobackの来日記念講演会で発表した。用いた原子団はJoback法の原子団をベースにしたので、JBDGC(Joback Base Dynamic Group Contribution)と名付けた。JBDGC法は、CH3がどのような原子団と結合するかによってCH3の数え方をダイナミックに変える。JBDGCは原子団寄与法の良いところと、結合情報の保持を併せ持つ方法である。この方法を使うと、pKaや誘電率など結合情報が物性に影響を与える物性の推算精度を精度よく予測することができる。改良系として原子団の拡張[*2]を図っている。Jobackの原子団とは離れていくので単にDGC法と呼ぶ。
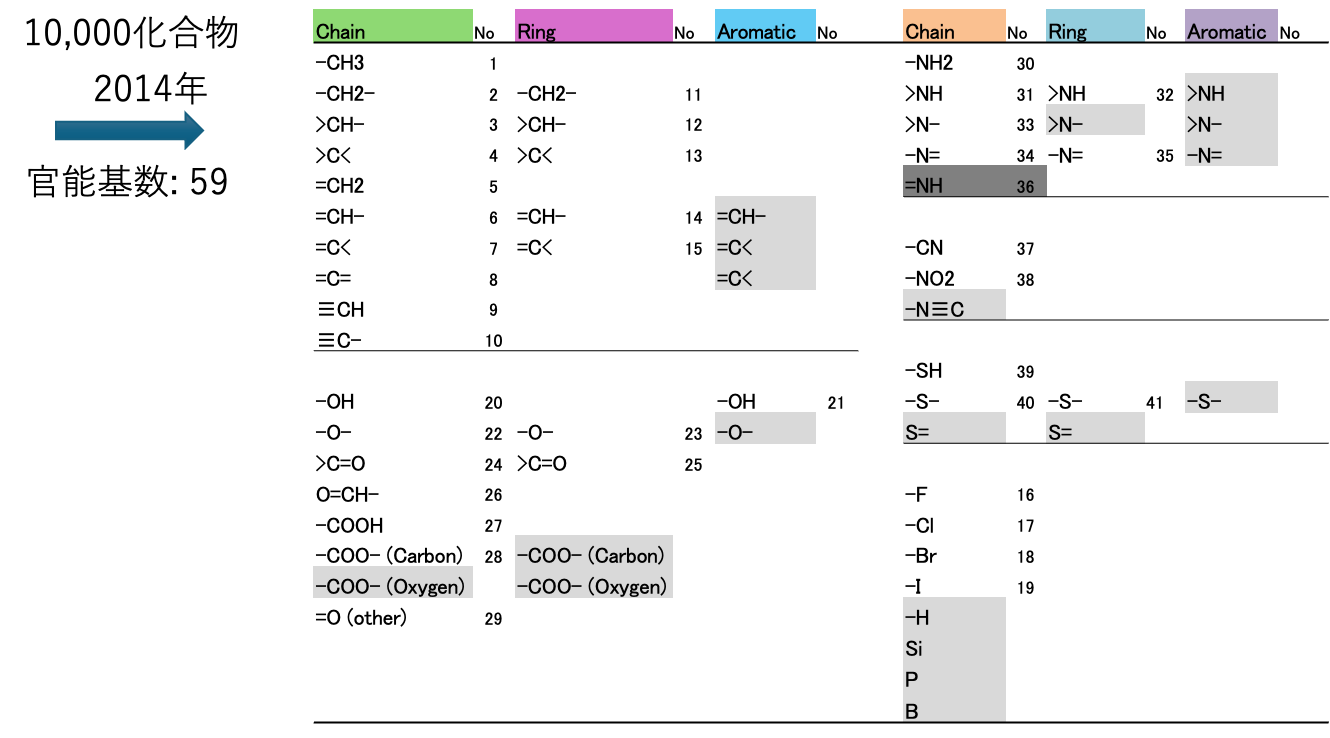
[2. DGC: Dynamic Group Contribution原子団リスト]
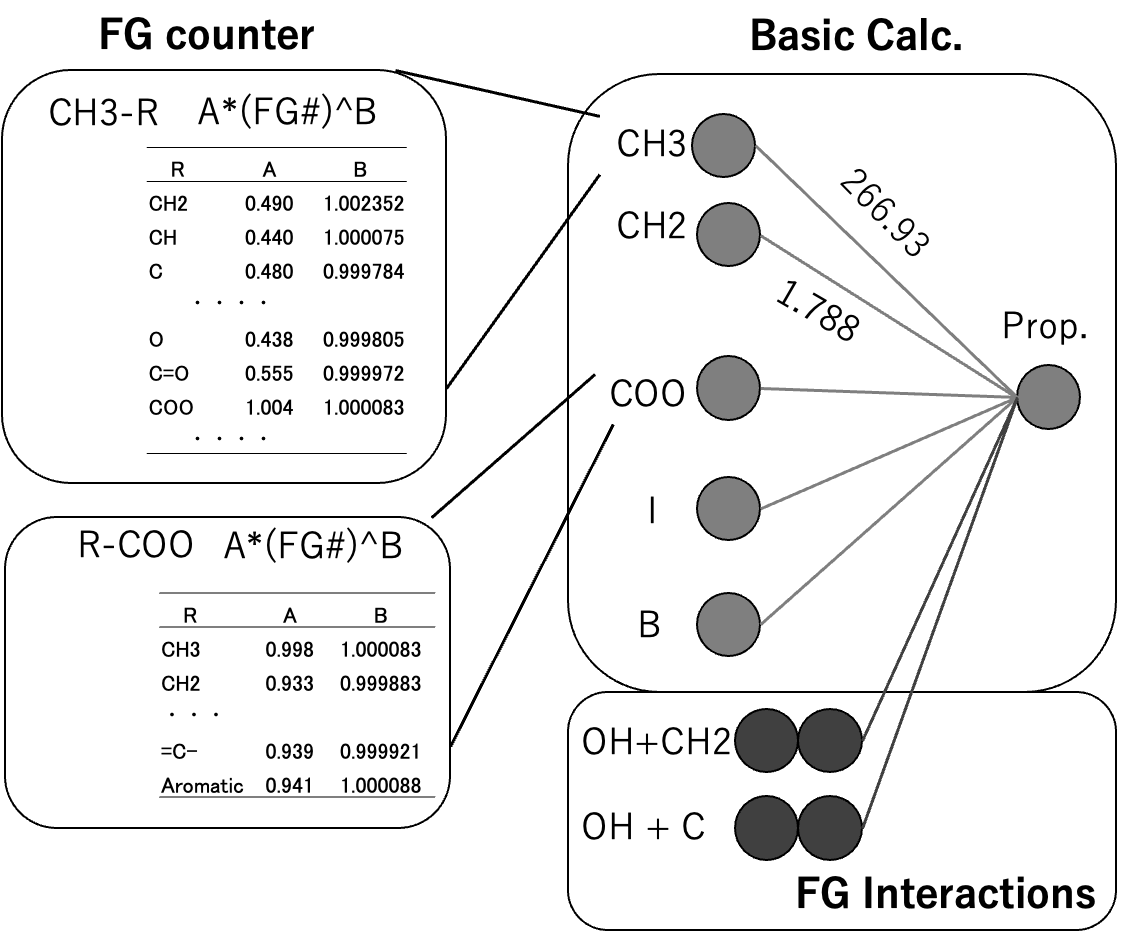
表1に2014年度版、DGC法原子団リストを示す。JOBACKの元の原子団に新たに19原子団(灰色)を加え、=NHを消去した。DGC用に、分子のSMILESの構造式[*3]をDGC法原子団リストに変化するプログラム、YMB4DGC[*4]を作成した。どんな有機化合物のデータセットを使うにしてもDCG2014年版ではこの59官能基を使う。実際には環の情報も使うので60だ。YMBの172官能基[*5]と比べるととても少ない官能基で物性推算することになる。
[2.1 DGC、Basicの計算]
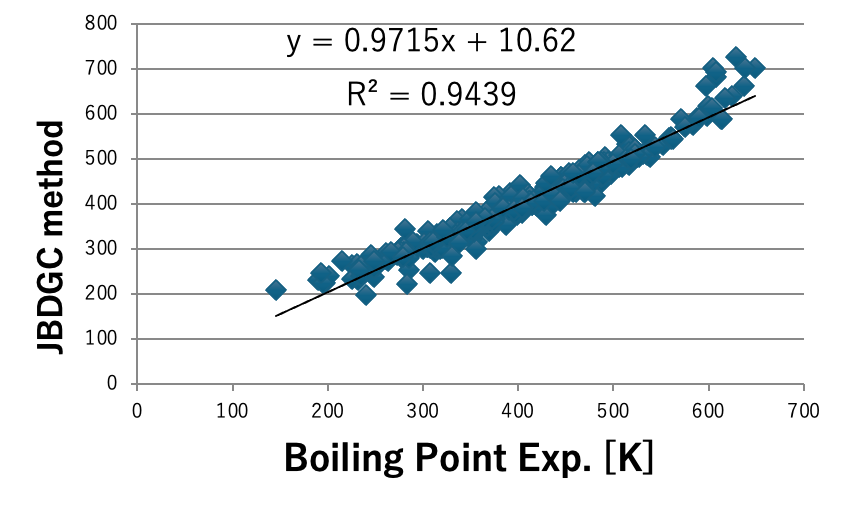
「The properties of Gases and Liquids 3rd 」という書籍には400化合物のデータが記載されている。これをメインの原子団の数だけから重回帰計算を行う。図1にDGC法ベーシックによる沸点の推算結果を示す。横軸は文献値の沸点、縦軸はDGCの計算値の沸点になる。このは単に拡張JOBACK法で計算した結果になる。定義されている原子団の数が少なければ表現できる分子も少ない。アミド基が定義されていないと、C=O(ケトン)とNH(アミン)で表現するしかないので推算精度は高くなり得ない。
[2.2 DGC計算の1番目のオプション]
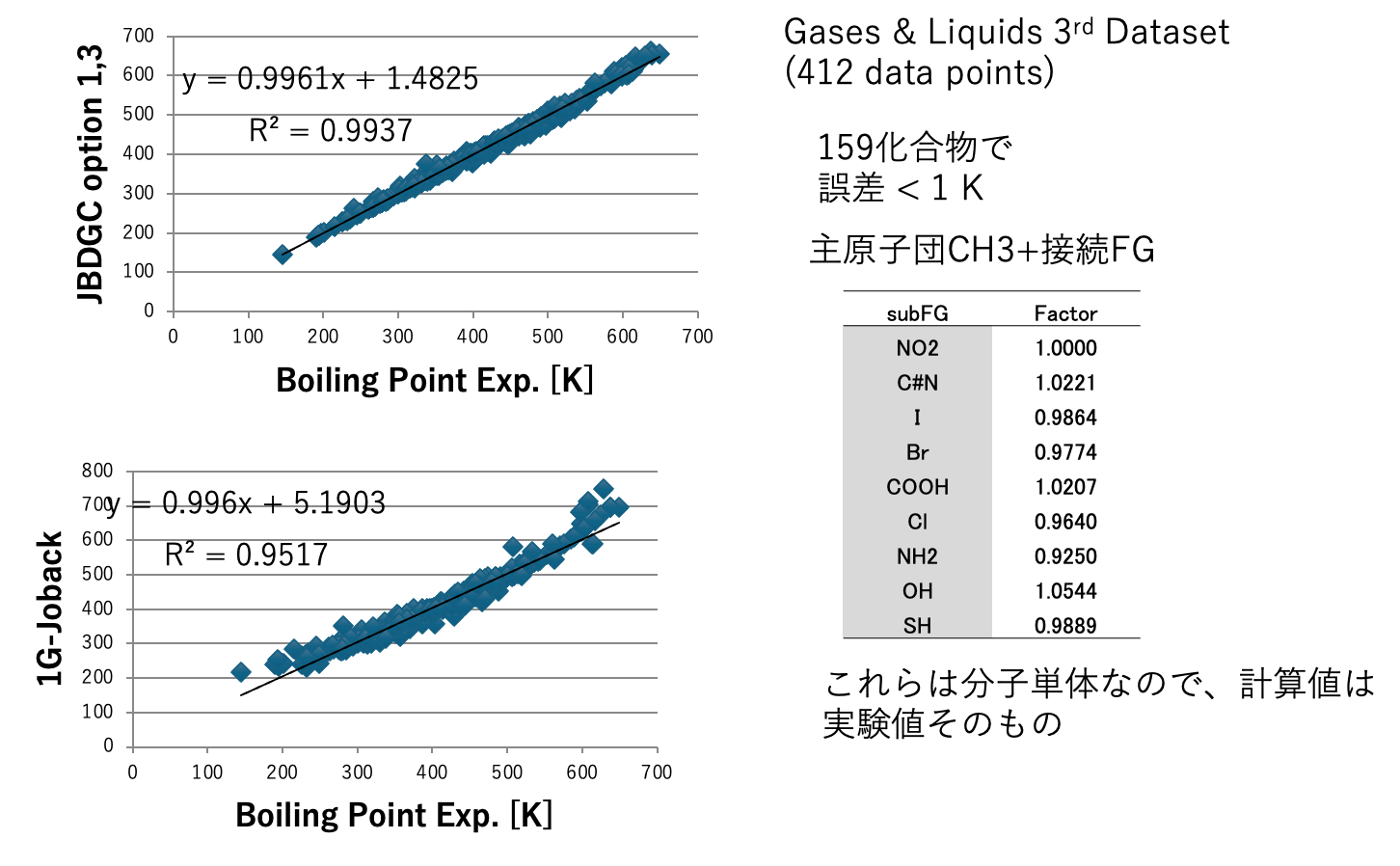
JBDGCでは、メインの59原子団がどんな原子団と接続しているかを調べる。表2に原子団の数え方を示す。メインの原子団がCH3の場合、CH3に何がつくかでCH3の数え方を変える。NO2, C#N, I, Br, O=CH, COOH, NH2, OH,SHの場合には分子そのものになる。例えばCH3NH2の場合、CH3は0.9250個と数える。結合相手、係数は解析したDataSetに依存する(Dynamicに変化してしまう)。数え方計算はGROVE法[*6]と同じく遺伝的アルゴリズム法(GA法)を用いる。教師データと計算値の誤差を偏微分係数に従って係数に戻す操作がないので、実験値に誤差がある場合に強い推算法になる。収束計算を行うので計算に時間がかかる。
[2.3 DGC計算の2番目のオプション]
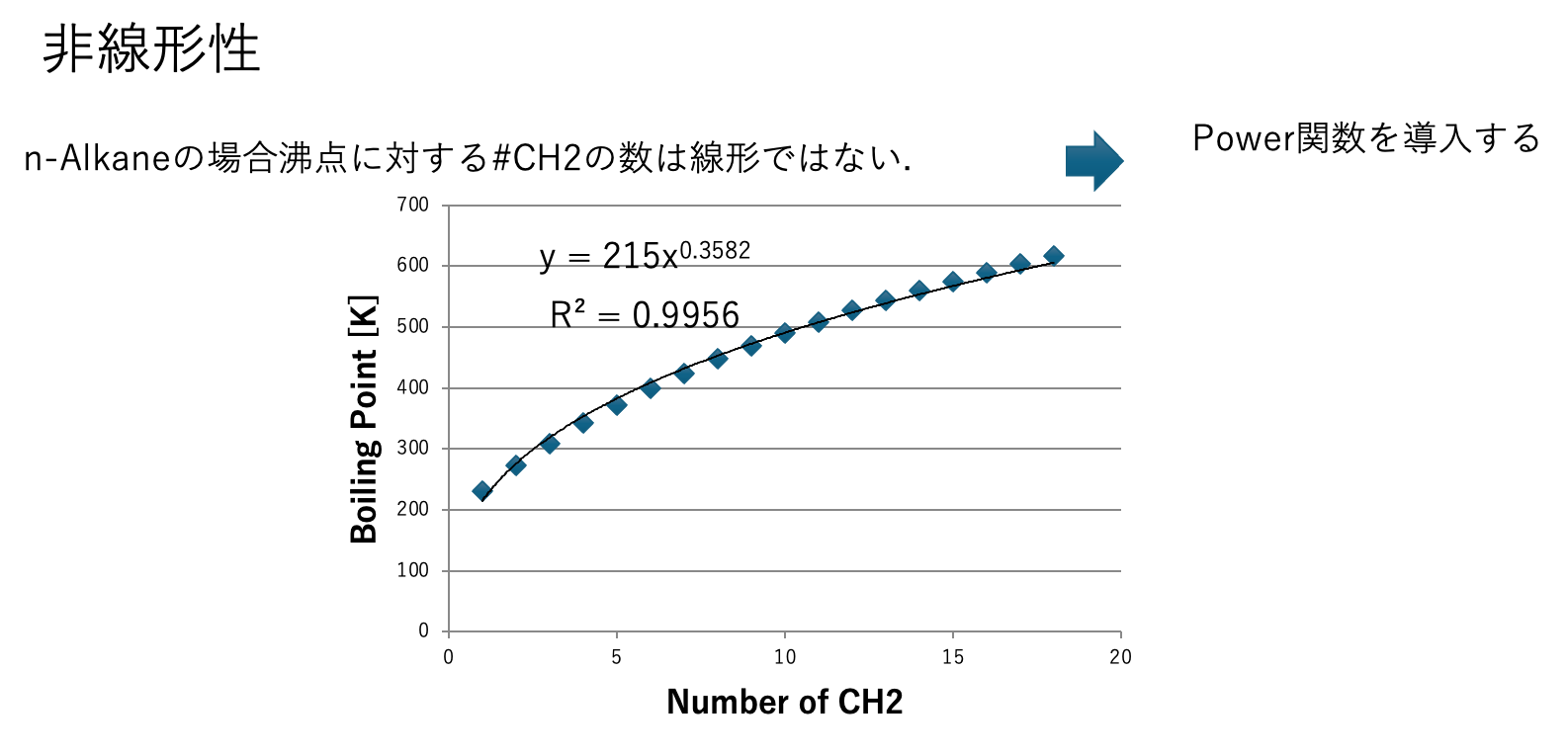
図2にn-アルカンの沸点を示す。メインCH2_subCH2_subCH2の数が増えると、沸点への影響が頭打ちになる事がわかる。メイン原子団のうち、分子を大きくする原子団はCH2, CH, C(環状を含む)である。他の原子団が分子中にたくさん入ることは少ない。そこで、オプション2が有効ななのは、炭化水素だけを大きさによって物性が変わるような系以外では効果がない。
[2.4 DGC計算の3番目のオプション]
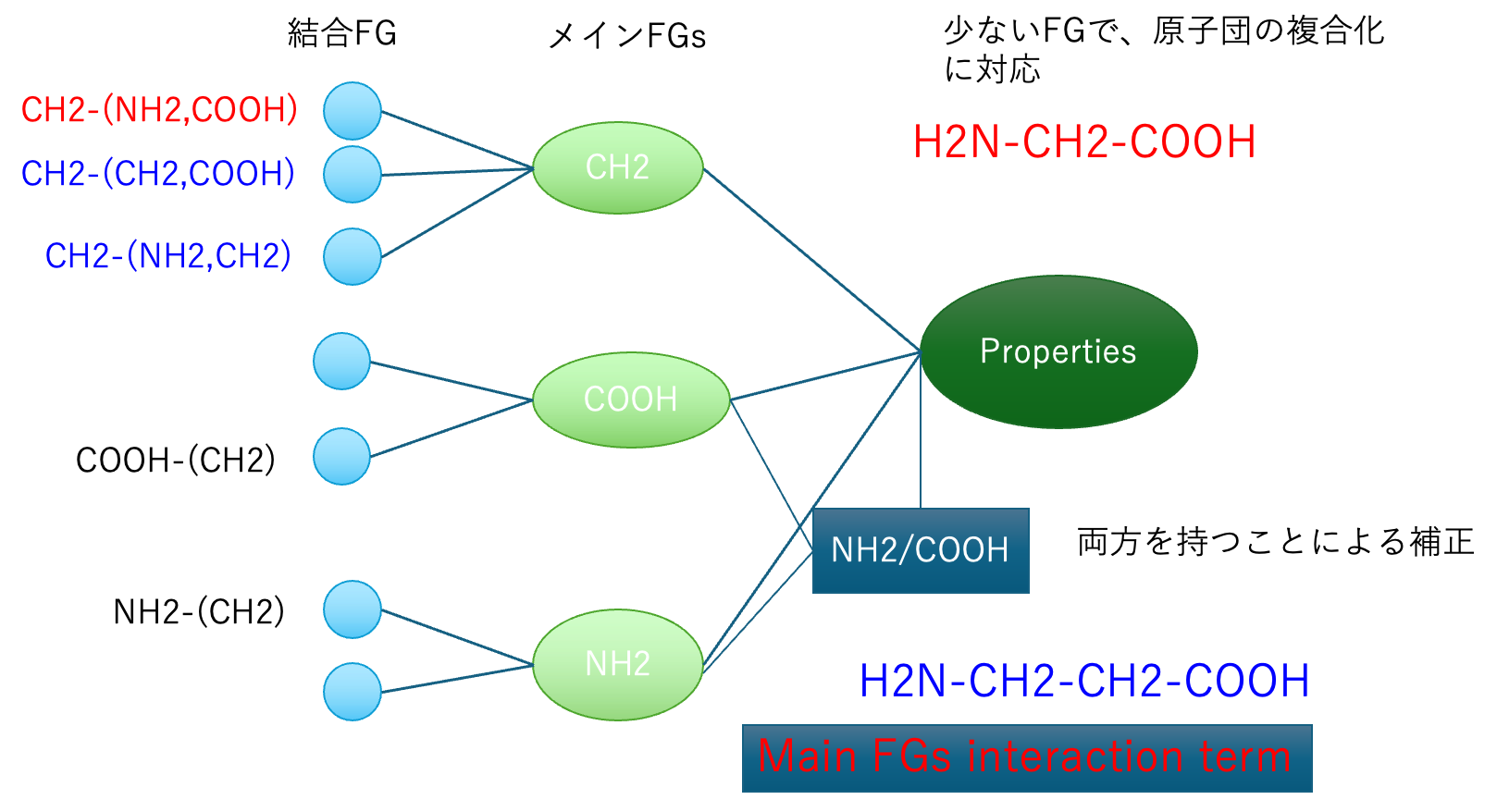
オプション2まで使いテーブルが作成されている。そのテーブルに対して、各メインFGが相互作用していると仮定した時に、どのようなFGペアの効果が高いか探索する。図3に示すように、分子中にCOOH基とアミノ基を両方持つ化合物の場合、様々な物性値に特異的ペアの影響が現れる。1つの炭素にカルボキシル基とアミンが付いたアミノ酸の場合には、CH_COOH_NH2の形でDGCで評価される。しかしCH2CH2基を介して両末端に付加した場合にはDGCでは評価にはいらない。そのような特異的ペアの数(クロスターム数と呼ぶ)を指定して大きな影響を与えるものをリストアップする。見かけ上の精度は高くなるが、あまり導入数を多くすると化学的な意味合いが薄れるので注意が必要である。現在のバージョン(2026)ではメタノール、CH3_OH, OH_CH3は1度しか使われないパラメータになる。そこで2番目のオプションで物性値予測はメタノールそのものになる。しかしクロスタームを導入すると、クロスターム分、実験値から外れる。
[2.5 DGC法の全体像]
図4にDGC法の全体像を示す。見ようによっては1種のニューラルネットワークになるがニューロンの結合を強く制限することによって予測性能が飛躍的に向上させ、過学習を抑制する。
[3. DGCのFitting性能]
一つの官能基は色々な分子で使われている。普通の原子団寄与法では、官能基の回帰係数は多くの分子での平均になる。結果として、例えばCH3(平均) + NH2(平均)のような単純な分子では誤差が大きくなる。DGCでは結合相手によって官能基の数え方を変える。CH3-NH2ではCH3は0.925個あると考える。そこでフィティング性能は非常に高くなる。例えば、CH3-CH3という化合物がデータセットに無い場合、subFGにCH3は無い。その場合、Factorは1.0を使う。最悪予測性能は第一世代のJOBACK法と同じになる。実験値が1つでもあれば、簡単にファクターを決めることができる
BP(184.55K)= 123.8928*Factor(CH3_CH3)*2+…..*0….*0
Factor(CH3_CH3)=0.7448
既に出来上がったものに付け加えても良いし、あらたに計算し直しても良い。
[4. DGCを使った実例]
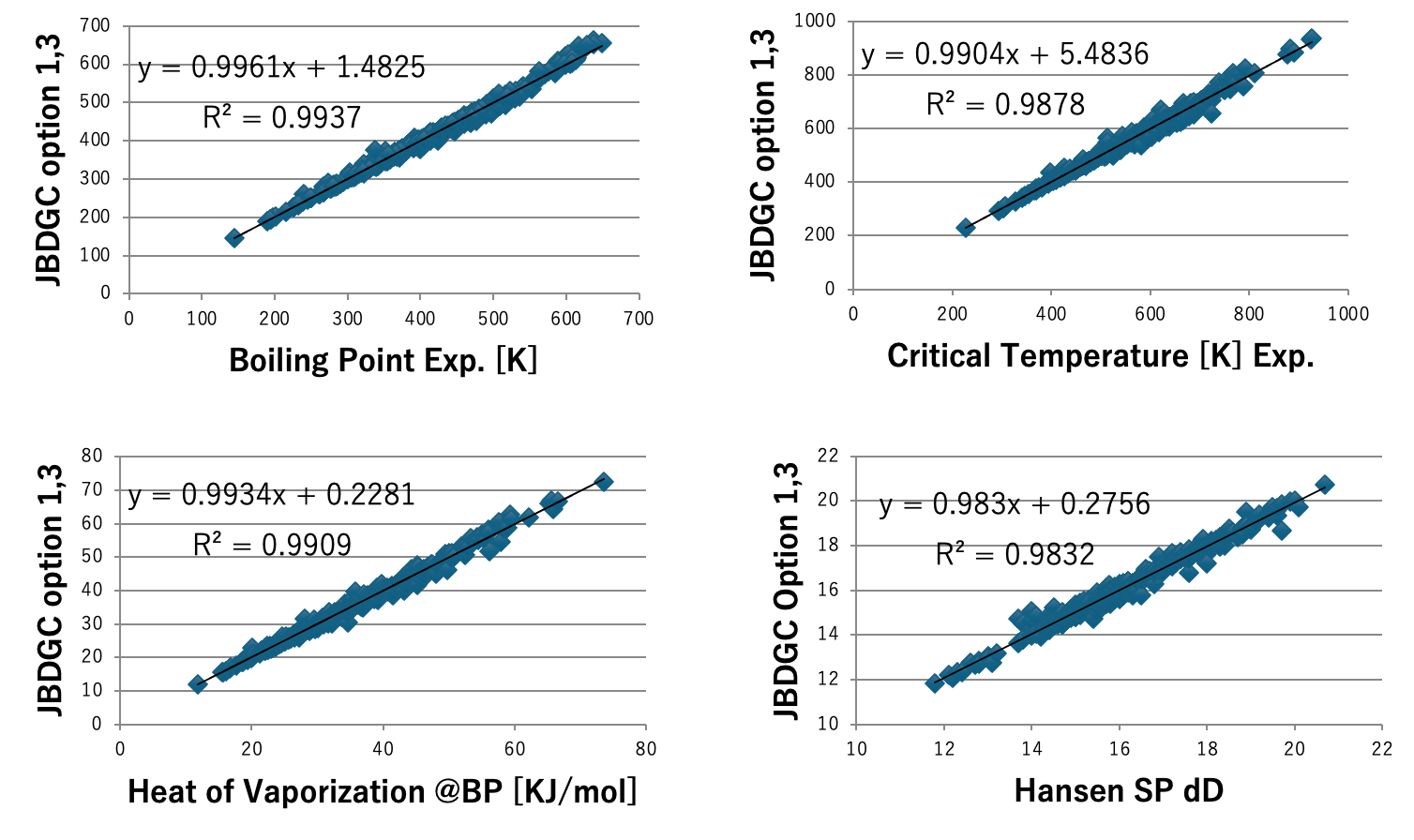
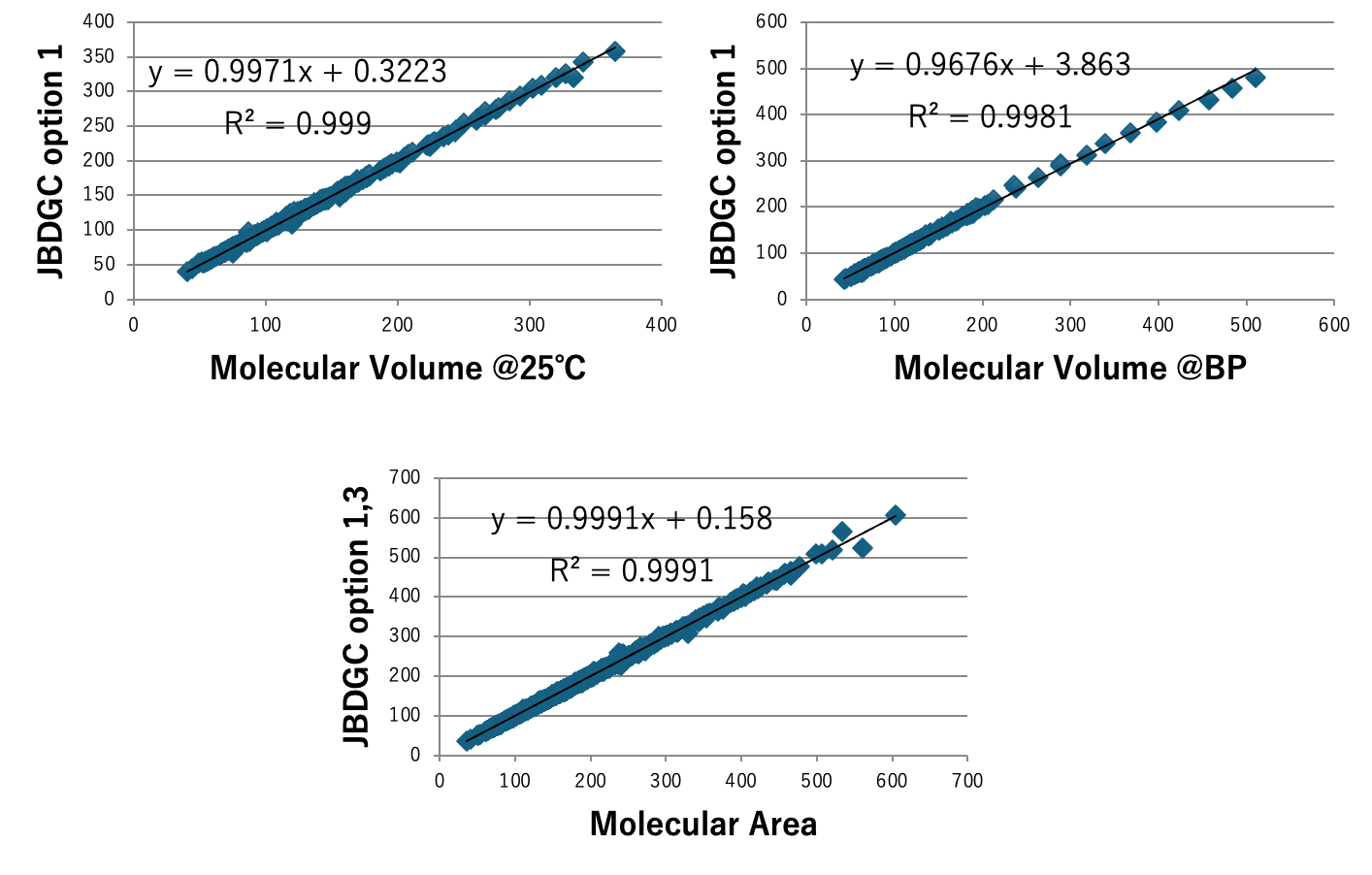
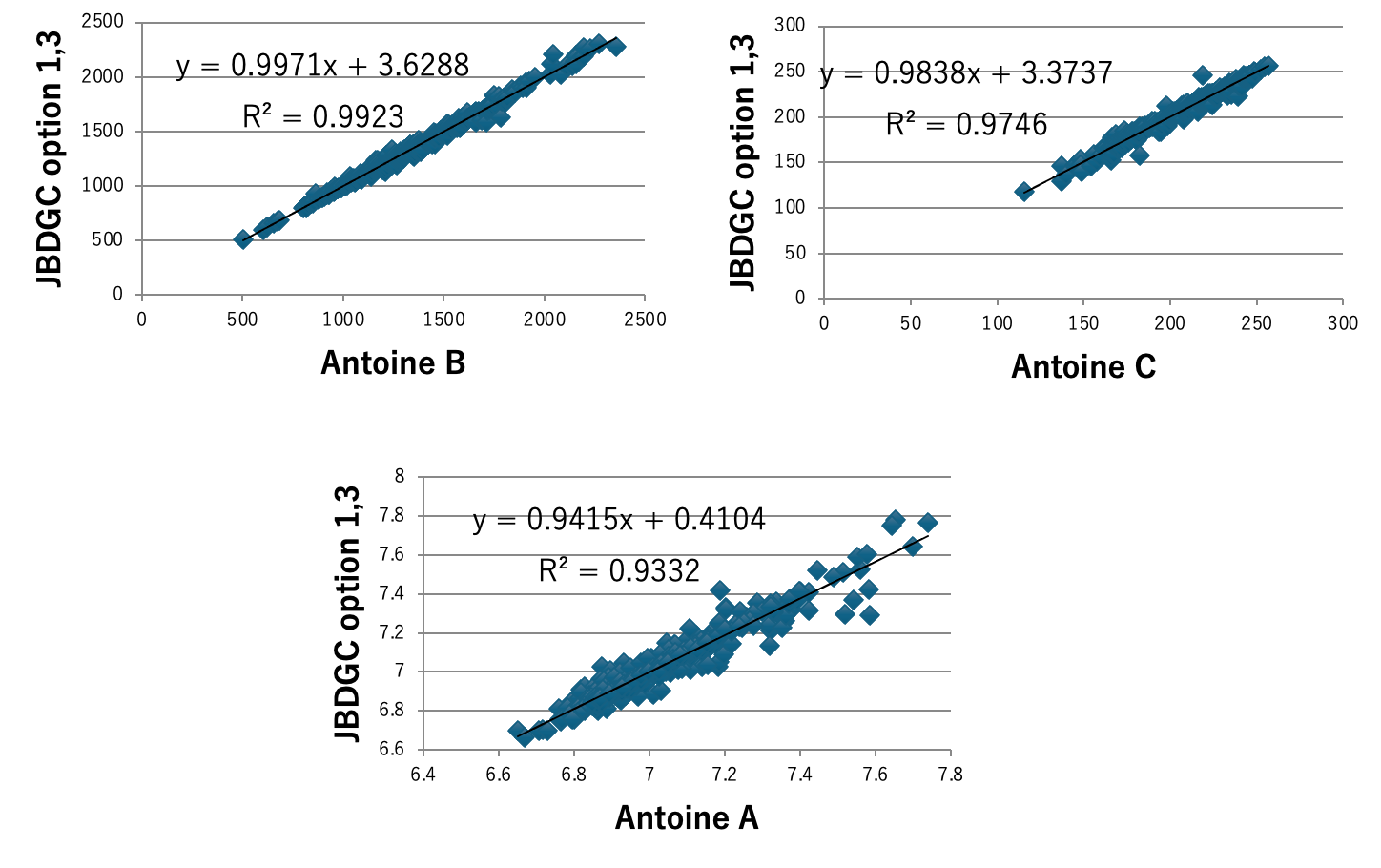
図5にDGC法で計算した沸点とJOBACK法で計算した沸点を示す。DGCでオプション1,3を加えると412化合物中159化合物で沸点誤差が1K以下になる。59官能基しか使っていないが、決定係数0.9937で沸点を推算することができる。ただし、内部的には各FGの結合情報によって原子団の個数の数え方を変えているので、ある意味官能基を1000種以上定義したのと変わらないかもしれない。沸点、臨界温度、沸点における蒸発潜熱、Hansenの分散項(dD)、25度での分子体積、沸点での分子体積、分子表面積、Antoine A, B,Cの推算式をDGC法で作成し図6-8に示す。
[5. DGCの利点と欠点]
利点:
- メインのFGsの数え方 = 知識になる
- 非常に多くの原子団を定義したのと同じ
- ニューラルネットワーク法と異なり係数の意味が明瞭.
- プログラマーには扱いやすい
- 自分のデータを取り込みやすい.
- 小さな重要な分子ほど精度が高い。
欠点
- プログラムサイズが大きくなる
- スプレッドシートで扱いにくい
- 計算時間がかかる
あるFGにあるFGが結合している。相手によってもとの原子団の個数の数え方が変わる。フッ素が付いたときはどうかわるか? それがヨウ素だったら? 水酸基だったら? 結果を見ているだけで楽しい。
[6. 図表]
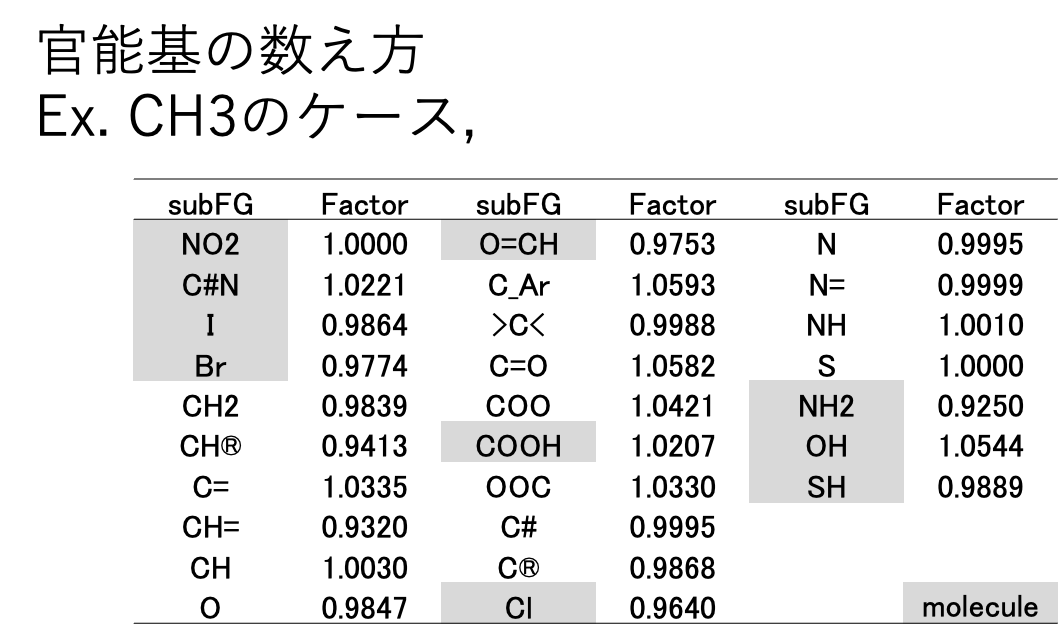
NO2, C#N, I, Br, O=CH, COOH, NH2, OH,SHの場合には分子そのものになる。
[7. Pirika.comへのリンク]
*1: Joback法
*2: DGCの原子団拡張
*3: SMILESの分子構造式
*4: YMB4DGC
*5: 官能基のリスト (TCPE), HSPiPで使える官能基一覧 (HSP/Docs)
*6: GROVE法
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)
メールの件名は[pirika]で始めてください。
