
2026.3.30
pirika.comで化学
>チャピエモン-3rd Pirika Origin (CPO)
> ハンセン溶解度パラメータ (HSP)
> 化学全般
> 情報化学 >物性化学 >高分子化学 >化学工学 >その他の化学 >昔のもの
>情報化学ツール >MAGICIAN養成講座 >STEAM
>Pirika ツール群
ブログ
業務案内
お問い合わせ
情報化学ツール > 原子団寄与法>
原子団寄与法の基礎
Dynamic Group Contribution
DGCの原子団拡張
DGCの応用例 > DGCを使ったAlfrey-PriceのQe値の推算
高分子化学 > ポリマー中のモノマーのシーケンス(並び方 > > Alfrey-Price Qe 値>
健忘症進むのは不幸せか? 同じようなブログがあってもお許しください。(Qe値推算ブログから転記)からQe推算Webアプリ(polymer)
[1.概要]
pirikaではラジカル重合のシミュレータにAlfrey-PriceのQe 値を使っている。次式で簡単に反応性比が得られる。
r1=Q1/Q2 ・e -e1(e1-e2)
r2=Q2/Q1 ・e-e2(e2-e1
このQe値を集めデータベース化[*1]した。集めたものをニューラルネットワーク法を用いて推算式を構築した。入力にはMOPACで計算したHOMO-LUMOの値や2重結合のHead-Tailの電荷などを入れる。その方法ではQe値が未知のモノマーに対しては分子軌道計算を行わなくてはならない。e値に関しては電荷と高い相関があるので比較的予測しやすい。ところがQ値に関しては2重結合の周りの環境に依存するためニューラルネットワーク法[*2]を用いると過学習[3]を起こしやすい。
[2. DGC法を用いたQ値の推算]
DGC法の使い方は「DGCの使い方」[*4]に従う。Smilesの構造式とQe値のデータのペアを用意する。多くの物性推算式の構築で共通のことであるが目的変数の値の範囲はよく考える必要がある。最大値と最小値が100倍異なるならlogを取った方が良い。ただし、最小値がマイナスの場合には配慮が必要になる。Q値の場合はゼロ以上なのでlogを取った方が良い。DGC結合テーブル作成用のYMB4DGC[*5]を使い表1に示す結合テーブルを作る。DGCプログラムで結合テーブルを読み込むとメイン原子団の数がリストアップされる。すぐに重回帰法[*6]で計算する。結果を図1に示す。この段階で大きく外れるものはデータが間違っている可能性がある。同じモノマーで複数のQe値がある場合にはより収束の良いデータと入れ替える。ここではデータのクレンジングは終了しているのでこのままDGCの計算に進む。表1の結合情報をもとにメイン原子団の数の数え方を変える。計算が終了すると表2に示すテーブルが作成される。そのテーブルから重回帰法でQ値を計算する。図2[b]に示すようにQ値はlogを取って解析した方が良い。計算が終わるとJavaScriptのプログラムが生成される。それを組み込むと分子構造からQ値を計算するWebアプリ(図3)になる。
[3. 原子団寄与法との違い]
例えばアクリル酸メチル(CH2=CH-COO-CH3)と酢酸ビニル(CH2=CH-OOC-CH3)は原子団の数は同じになる。アクリル酸メチルのQ値は0.42、酢酸ビニルのQ値は0.026と大きく異なる。DGC法では、CH2=につくのは=CHなので同じになる。しかし=CHにCOO(炭素の側が付加)場合には=CHは1.252個あると考える。=CHにOOC(酸素の側が付加)場合には=CHは0.585個あると考える。CH3の場合相手によってCOO(0.787個)OOC(0.747個)と数える。アクリル酸メチルのQ値の予測値は0.40、酢酸ビニルのQ値予測値は0.044と大きく改善される。
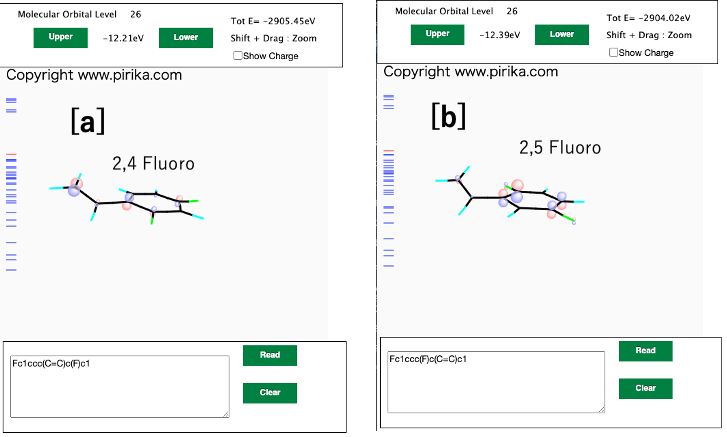
DGC法を用いてもダメなものはある。図2[b]で精度の出ないものをみると、2,4-Difluorostyrene (Q=0.65)、2,5-Difluorostyrene(Q=6.7)と大きく異なるが、DGC法ではどちらも1.825になってしまう。ベンゼン環は電子を引っ張るフッ素がついた場合にo-, p-とo-, m-で電子の引っ張られ方が大きく異なる。そうした効果を入れるためにはDGC法の改良が必要である。CNDO/2[*7]で計算すると図4に示すようにスチレンTailのπ軌道の大きさが大きく異なる。これがQ値の違いに寄与している可能性がある。
[4. 図表]
| Name | logQ | b | b | b | b | b | b |
| Acrylamide | 0.0492 | 5_6 | 6_21_5 | 21_33_6 | 33_21 | ||
| Acrylic acid | 0.1038 | 5_6 | 6_23_5 | 23_6 | |||
| Acrylonotrile | -0.2218 | 5_6 | 6_43_5 | 43_6 | |||
| Allylacetate | -0.6198 | 1_24 | 24_31_1 | 31_24_2 | 2_31_6 | 6_5_2 | 5_6 |
| ・・・・・ | ・・ | ・・ | ・・ | ・・ | |||
| N-octylAA | -0.7212 | 1_2 | 2_2_1 | 2_2_2 | 2_2_2 | 2_2_2 | 2_2_2 |
| N-PhenylMA | -0.0706 | 17_34_16_16 | 16_17_16 | 16_16_16 | 16_16_16 | 16_16_16 | 16_17_16 |
| DiethylaminoEMA | 0.3181 | 5_7 | 7_24_5_1 | 1_7 | 24_31_7 | 31_24_3 | 3_35_31_1 |

| ID | Prop | CH3 | CH2 | CH | C | CH2= | CH= | C= |
| 23 | 0.049 | 0 | 0 | 0 | 0 | 1.094 | 0.994 | 0 |
| 24 | 0.104 | 0 | 0 | 0 | 0 | 1.094 | 0.991 | 0 |
| 25 | -0.222 | 0 | 0 | 0 | 0 | 1.094 | 0.987 | 0 |
| 27 | -0.620 | 0.787 | 0.564 | 0 | 0 | 1.094 | 0.947 | 0 |
| ・・ | ・・ | ・・ | ||||||
| 20628 | -0.721 | 0.974 | 0.783 | 0 | 0 | 1.094 | 0.994 | 0 |
| 20629 | -0.071 | 0 | 0 | 0 | 0 | 1.067 | 0 | 0.901 |
| 21195 | 0.318 | 3.790 | 2.001 | 1.279 | 0 | 1.067 | 0 | 1.029 |

[5. pirika.comへのリンク]
*1: Qe 値の推算 2005.7.20
*2: ニューラルネットワーク法
*3: 過学習
*4: DGCの使い方
*5: DGC結合テーブル作成用のYMB4DGC
*6: 重回帰法
*7: 自作のCNDO/2
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)
メールの件名は[pirika]で始めてください。