
2026.3.7改定 (2003年頃の記事)
pirika.comで化学
>チャピエモン-3rd Pirika Origin (CPO)
> ハンセン溶解度パラメータ (HSP)
> 化学全般
> 情報化学 >物性化学 >高分子化学 >化学工学 >その他の化学 >昔のもの
>情報化学ツール >MAGICIAN養成講座 >STEAM
>Pirika ツール群
ブログ
業務案内
お問い合わせ
情報化学 > 情報化学ツール > 過学習と予測性の欠如
[1. 概要]
ニューラルネットワーク法[*1]は非線形の解法である。非線形性に追随するためにデータ点周辺で大きく値を変化させてしまう事がある。データ数が少なすぎる、中間ニューロンが多すぎる、収束の閾値が小さすぎる時に起こる。イメージ的には、データ点をなめらかに繋ぐ曲線を探し出してしまう。
その現象を過学習という。見かけ上の相関係数はとても高く、高精度なモデル式が作れたように勘違いする事が多い。しかし学習点を少し外れるだけで、値は大きく変化するので実用上の予測性能はほとんどない。作成されたNN法を評価するときにデータの一部を学習させずに予測データとして残しておく事が行われる(Leave several out法)。
山本のモデルの確認法は、適当なデータ点の周辺を計算し、値が大きく変動するかどうか確認することだ。
[2. NN法の過学習]
コンピュータは人間と違い、飽きることを知らない。命令すれば一晩中でも、1年でも学習を続ける。
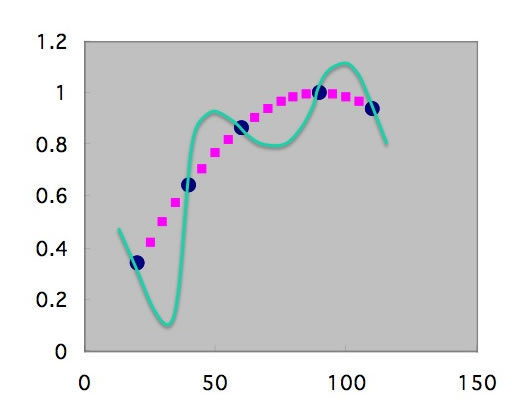
その結果、本来欲しい点はピンクの点であるのに、最終的には学習した点をなめらかにつなぐ緑の線を答えとして提案する。学習に使ったデータでは相関係数R=1.000となる。このモデルは学習した近傍以外は非常に悪い答えを返す。これなら、重回帰のほうがまだましというレベルだ。
このように単純な系では、間違いはすぐにわかるが、複雑な系では、過学習が起きたかどうかは非常に分かりにくい。過学習は、学習のデータポイントが少ない、NNの中間ニューロン数が多すぎるときに起こりやすい。そこで、ピンクの点も含めて学習させてしまうことを考える。
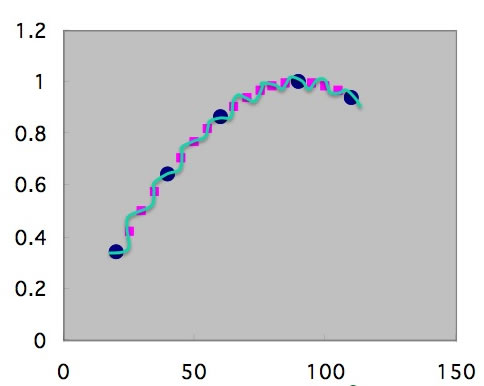
データ数が増えてくれば大きくハズレる曲線は取りようがなくなる。
これが、機械学習にはビッグ・データが必要という事につながる。
中間層のニューロン数は次式でシグモイド関数を何個足して、この現象を表現しようとするかで決まる。
f(y)= A1* / [1+exp(-(B1*X1+B2*1))+
A2* / [1+exp(-(B3*X1+B4*1))+
A3* / [1+exp(-(B5*X1+B6*1))
ニューロン数を増やせば複雑な現象にも追随できるが、その分、過学習という、やっかいも背負いこむ事になる。中間層のニューロン数の最適化とデータポイントの数、これが考慮されずにニューラルネットワークを組んでも、無駄になる。
それを評価するのに赤池情報量基準(AIC)というものも知られているが、難しすぎて山本には理解出来できていない。
元々化学の現象はそれ以前に多くの、誤差、あやふやさを含んでいる。
[3. 予測性の欠如]
Sinカーブの学習で、Sinの頂点までしか学習しなかったとする。
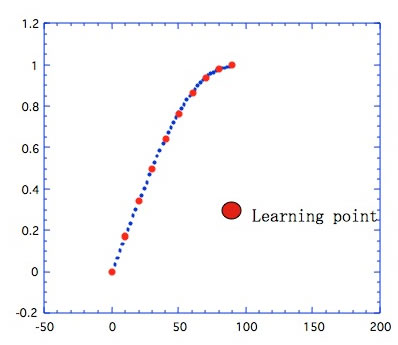
学習した内側については、中間ニューロンの数が適当であれば、図3の青い点が示すように良好に予測することができる。これは、”内挿問題に対する予測性”は高いという言い方になる。
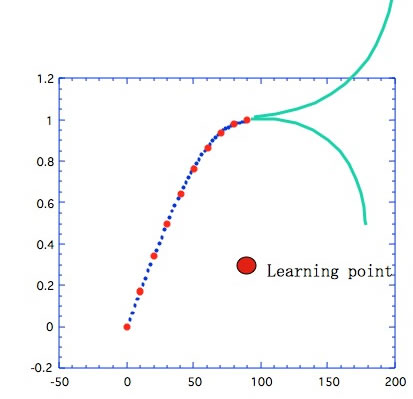
しかし、図4に示すようにθが90を超えた先がどこに行くのかはNNには判らない。(人間にも解からない)したがって1点でもよいから90度以上のデータを入れる必要がある。これは実は先程の過学習よりも、よっぽどたちが悪い事だ。
例えば、化合物の官能基を定義して沸点を推算するNNを構築しようとする。
ある官能基、例えば水酸基を持つ化合物の沸点のデータが水酸基が1,2,3個もつ化合物を含む沸点データがあり、NNに学習させたとする。分子中に水酸基を4つ持つものは、”外挿” 問題になり、予測性は高くないだろうと見当がつく。ところが、カルボン酸を1-2個もつ化合物、アミノ基を1-2個持つ化合物をNNに学習させる。分子中に、カルボン酸とアミノ基を両方もつアミノ酸をNNに予測させようとすると、これはやはり外挿になってしまう。こうした”組み合わせ”外挿はどれだけあるのかは、一般的に言えないし、非常にむずかしい問題だ。また、アミノ酸のように、ひとつの炭素にカルボキシル基とアミノ基が付いている場合、遠く離れて付いている場合でも物性値は大きく異なる。
ガラスの物性推算であっても、どの成分が主のドメインを作るかによって、学習していないものは外挿になりうる。
従って、NNで物性推算式を構築した場合には、どのような母集団からNNを構築したのかが非常に重要な問題になる。
[4. クロスバリデーション(交差検証)]
数学的な意味合いでクロスバリデーションの結果は論文を書く時に問われる。Leave several out法を使うケースが多い。しかし多くの論文を見ると、結構ずるいことをしているようにも見える。先ほどの例で、アミノ酸のデータが、ある酸化物が1度しか使われていないデータセットではアミノ酸やある酸化物を検証用に使ったら必ず悪い結果しか出ない。複合酸化物で例えばチタン酸バリウムは組成がぴったりの時に特異的な強誘電率[*2]になる。他のガラス組成の物性値とクロスバリデーションをしても意味がない。
非常に多くのデータがあり外挿性がない状況でのクロスバリデーションは意味があるかもしれない。しかし、毒性の場合悲しいくらいのデータしかない。将来iPS細胞を使ってハイスループットで毒性の評価値が得られるようになるまで待つしかない。
有機化合物の水への溶解度推算[*3]ではデータセットが大きくなるにつれ、前のCVは何だったのか?と言うくらい推算精度が悪くなる。
結局外挿問題が解決できないため、アルコール化合物に限る、ホウ珪酸ガラスに限ると内装問題にする。するとビッグデータにならない。これを延々と繰り返しているように見える。化学の問題にニューラルネットワーク法を適用するのは難しい。物理の問題のように法則と言えるものができあがれば、AIも困らないだろう。
[5. pirikaの検証法]
山本はこれまでに多くの物性推算式を構築してきた。その推算式はHSPiP[*4]やPirika Pro[*5]に搭載して販売されている。その際に一番注意するのはデータポイント周辺での値の変化である。
ガラスの誘電率をpirikaの持つ複数の非線形推算式で解析した。
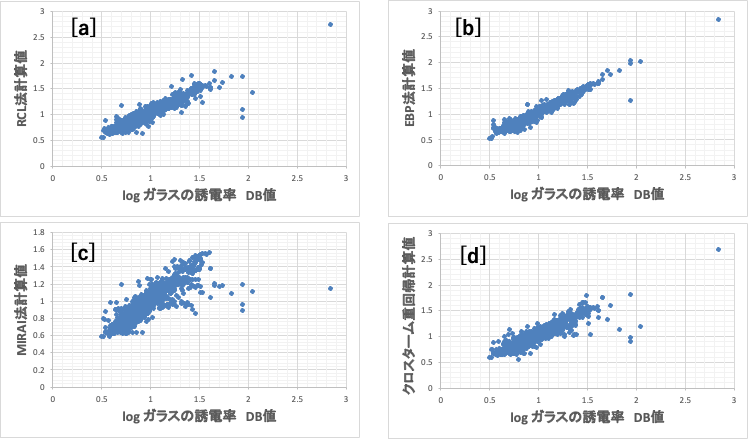
[a]再構築学習(RCL)ニューラルネットワーク法[*6]、
[b]誤差逆伝播NN法[*1]、
[c]MIRAI法[*7]、
[d]クロスターム重回帰法[*8]
ガラスの誘電率の計算精度の観点からは[b]の誤差逆伝播法が一番高い。
この4つの推算式を使って適当なデータポイント周辺を計算してみる。
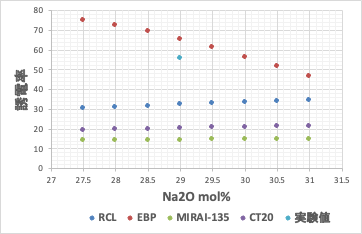
実験値は[b]誤差逆伝播NN法で計算したものに一番近い。ところがNa2Oを29mol%から増減すると[b]では計算値が46.7-75.1と大きく変化してしまう。シンプルなEBP-NN法で推算式を構築すると過学習を起こしている事が確認される。
過学習を起こし予測性が欠如している推算式を用いた場合には、組成の逆設計ができない。つまり、[b]式を用いて、より高い誘電率、より低い誘電率のガラスを設計しようとする。実験値のすぐ周辺に欲しい答えがあると認識してしまう。
これが化学系でDXが進まない大きな原因である。
[5. pirika内リンク]
*1 ニューラルネットワーク法
*2 ガラスの誘電率の推算法
*3 水への溶解度
*4 Hansen Solubility Parameters in Practice
*5 Pirika Pro
*6 再構築学習ニューラルネットワーク法
*7 MIRAI法
*8 クロスターム重回帰法
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください) メールの件名は[pirika]で始めてください。