
pirika.comで化学
>チャピエモン-3rd Pirika Origin (CPO)
> ハンセン溶解度パラメータ (HSP)
> 化学全般
> 情報化学 >物性化学 >高分子化学 >化学工学 >その他の化学 >昔のもの
>情報化学ツール >MAGICIAN養成講座 >STEAM
>Pirika ツール群
ブログ
業務案内
お問い合わせ
MAGICIAN養成講座 > pirikaのやり方 > ガラスの誘電率推算法
[1. 概要]
ガラスの誘電率と組成のmol%のデータ・セットが2443種ある。原料は61種類の酸化物、フッ化物だ。データの1次クレンジングは既に済んでいる(2つの原料はデータセットに無くなった)。研究の目的は誘電率をガラス組成から推算する式を構築し、任意の誘電率を持つガラス組成を逆設計する事だ。触媒設計や固体電解質の処方設計などにも使える重要な技術だ。
[2. 過去の技術]
Appenのガラス物性値推算法(1970年)が知られている。39種類の原料が定義され、19種類の原料に対して誘電率のファクターが決められている。
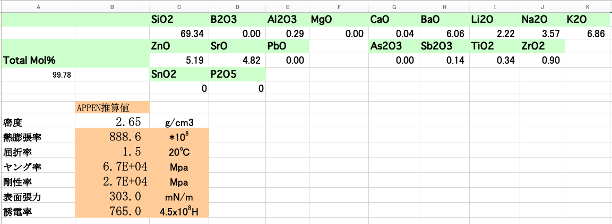
各原料の加算因子が決定してあれば組成がわかれば物性値を計算できる。Excelのスプレッドシートも図1に示すように簡単に構築できる。今回のデータセットを用いて、Appen式を拡張する方法は別ページで解説[*1]する。
[3. NN法を用いた誘電率推算]
再構築学習法[*2]ニューラルネットワークに誘電率を学習させる。
計算が進むと結合荷重が大きなものはますます大きく、小さなものはますます小さくなりメリハリが出てくる。中間ニューロンの孤立化も進み初期のニューロン30に対し16まで減った。
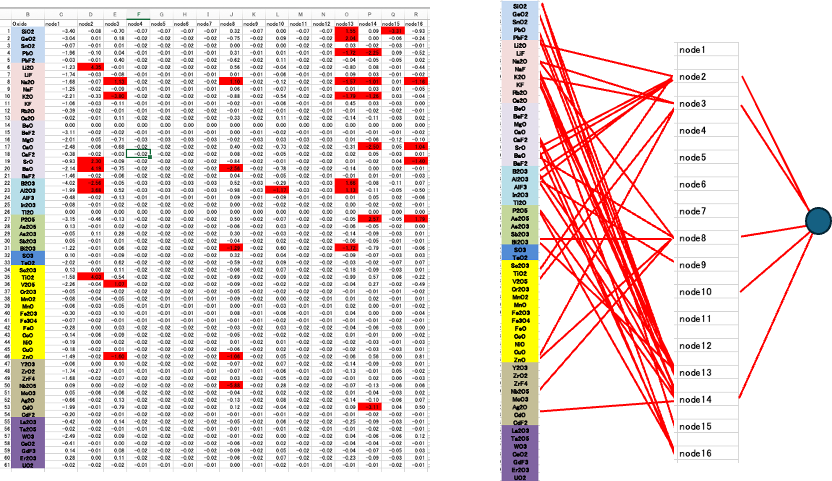
結合荷重の大きなものだけ見ると情報の流れが明らかになってくる。絶対値で1以上のものを赤くマークした。それ以外の結合荷重はほとんどが0.02程度なので誘電率にはあまり寄与しない。
通常の誤差逆伝播法のNN[*3]で同じように計算してみると、結合荷重の絶対値が0.02以下になるものは非常に少ないことが図3からわかる。
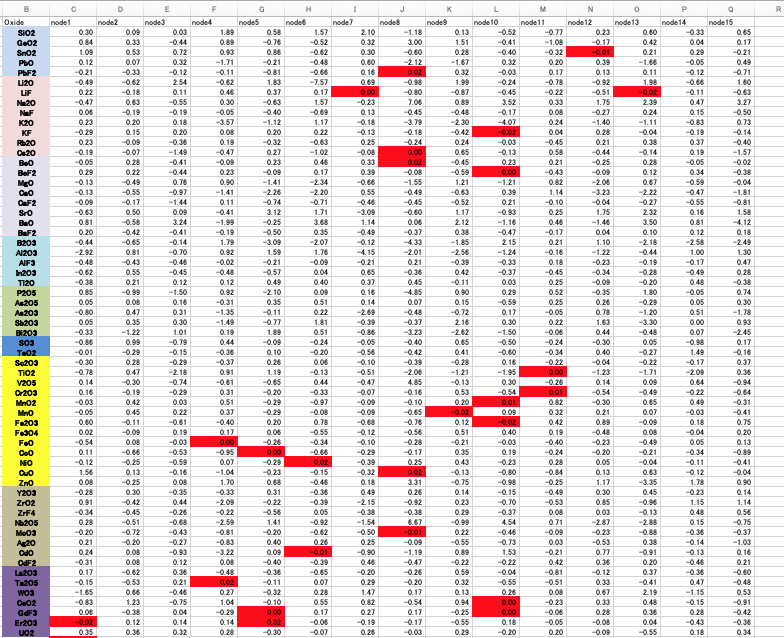
そこでNN法は中身がブラックボックスで情報を追いかける事ができないと言われてきた。例えばTiO2とLi2Oは強い相互作用をすることが分かっている。それでは同じように強く相互作用するペアを探索したい場合に、結合荷重行列からは何もわからない。
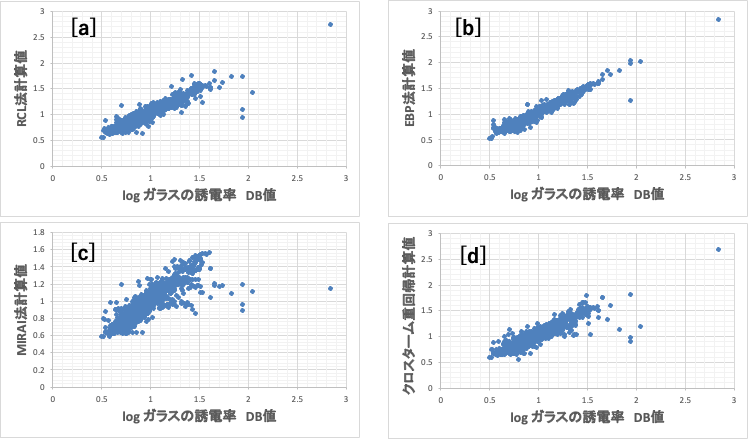
学習に使ったDB値と計算値の比較を図4に示す。[b]クラッシクなNN法を用いた方が精度が高いように見える。これは数値解析法の根源的な問題であるが、教師データは正しいと解釈するしかないのでこのような結果になる。おかしなデータであっても結合荷重を微妙に調整し線の上に乗せてしまう。その結果、そのデータポイントの周辺では少し値が変わるだけで大きく値が変わってしまう過学習[*4]が起こる。そして予測性能の低下が起こる。
再構築学習法では結合荷重の自由度が下がってしまうために、見かけ上の記述性は高くない。
[4. 情報の流れの検証]
再構築学習法では、図2の右側のニューロン同士の結合図に示す情報の流れが得られる。重回帰法[*5]は式の形から情報の流れはとてもすっきりしている。重回帰法にクロスタームの形で非線形性を導入する[*6]。情報の流れを重回帰のままに、非線形性の効果を見る事ができる。
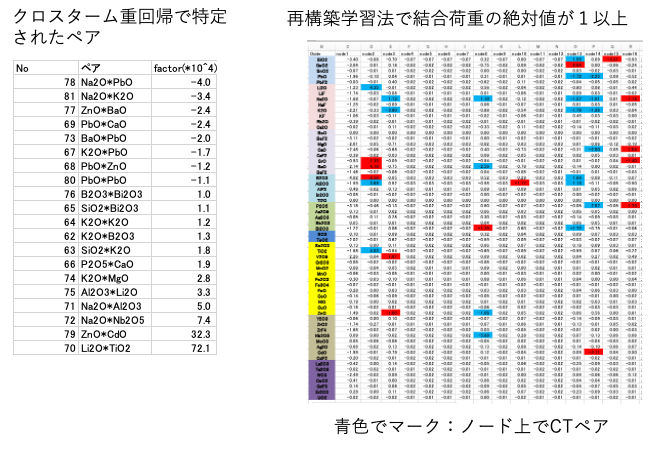
クロスタームで選ばれた酸化物ペアの意味合いは明確だ。あるペアが存在した時に、元の酸化物のファクター以上に誘電率の値を増減させる。その非線形性が再構築学習法の結合荷重に現れるとしたらどのような情報の流れになるだろうか?
例えばLi2OとTiO2は大きなクロスターム係数をもつ。結合荷重でのnode2のLi2OとTiO2は大きな結合荷重を持つ。node2ではAl2O3も大きな結合荷重を持つ。Li2OとAl2O3は大きなクロスターム係数を持つ。
このように大きなクロスターム係数と中間層ノード上での大きな結合荷重がリンクしているものは情報の流れが非常に明確になる。図4の[a]再構築学習法と[d]クロスターム重回帰法の相関が似ているのは偶然ではない。情報の流れが似ていることに起因している。
[5. モデルの検証]
過学習と予測性の欠如[*4]で説明したが、山本はクロスバリデーション検証はあまり行わない。結果が良くてもモデルが良いとは言えない。外挿データが増えたらCV評価は簡単に覆されてしまう。pirikaの評価は学習データポイントの周辺を評価[*4]することで行う。
図4で4つの非線形の方法でガラスの誘電率の推算式を作成した。
[a]再構築学習(RCL)ニューラルネットワーク法[*2]、
[b]誤差逆伝播NN法[*3]、
[c]MIRAI法[*7]、
[d]クロスターム重回帰法[*6]
ガラスの誘電率の計算精度の観点からは[b]の誤差逆伝播法が一番高い。
この4つの推算式を使って適当なデータポイント周辺を計算してみる。
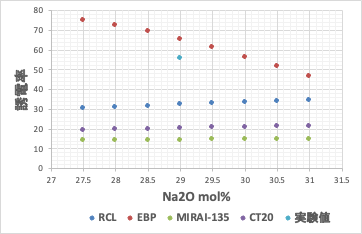
実験値は[b]誤差逆伝播NN法で計算したものに一番近い。ところがNa2Oを29mol%から増減すると[b]では計算値が46.7-75.1と大きく変化してしまう。シンプルなEBP-NN法で推算式を構築すると過学習を起こしている事が確認される。
過学習を起こし予測性が欠如している推算式を用いた場合には、組成の逆設計ができない。つまり、[b]式を用いて、より高い誘電率、より低い誘電率のガラスを設計しようとする。実験値のすぐ周辺に欲しい答えがあると認識してしまう。
これが化学系でDXが進まない大きな原因である。
[a],[c],[d]ではNa2Oの量を変化させても誘電率は大きく変化しない。どのモデルが良いのか、どれだけデータポイントをチェックすれば検証したことになるのか難しい。しかし情報の流れを検証する事ができる。非線形性の大きなデータポイントから調べれば良い。
[6. MIRAI法]
MIRAI法[*7]も非線形式であるが、図4[c]に示すように[a][b]とは少し異なる挙動を示す。
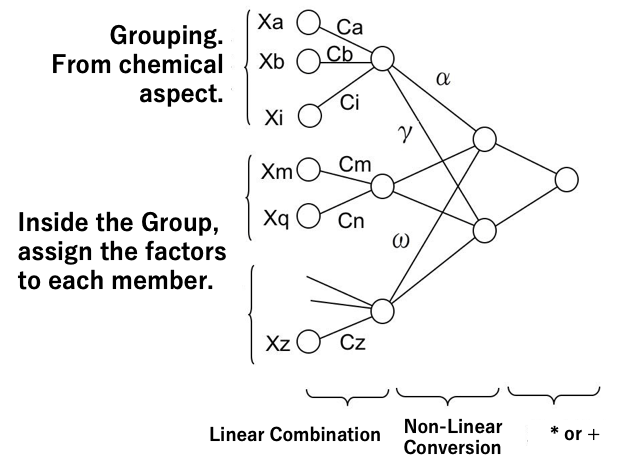
MIRAI法は入力ニューロンをグループ分けする。そしてグループ内では線形結合する。グループの頭の中間ニューロン同士は非線形結合する。結合荷重を刈り込み処理しないクラッシクなニューラルネットワーク法[*3]、再構築法でニューロンの孤立化を図る再構築学習法NN[*2]と比べグループを人間の研究者が指定することでネットワークの単純化が図られる。
図4を見る限り、相関係数は一番低いように見える。
その理由はネットワークの単純化だけのせいではない。MIRAI法だけfeed forward法を使っている。重回帰法の最小二乗法も誤差逆伝播法も教師データが正しいと考える。そして誤差を重回帰係数や結合荷重に落とし込む。誤差の多い系では、見かけの相関係数は小さいが、予測性能では逆に高くなる。
ここで問題なのは、TiO2とLi2Oが相互作用する図4の一番右の点である。MIRAI法[c]以外は計算値が高くなる。TiO2とLi2OはMIRAI法のグループが異なるので直接相互作用をできないので当然の結果だ。
そのような挙動を示す他のパターンをどう探す事ができるか?
これが問題だ。
MIRAI法の結果を示す図4[c]で、縦軸(MIRAI法計算値)が同じなのに、横軸(誘電率のDB値)が大きく異なるデータを抽出する事ができる。
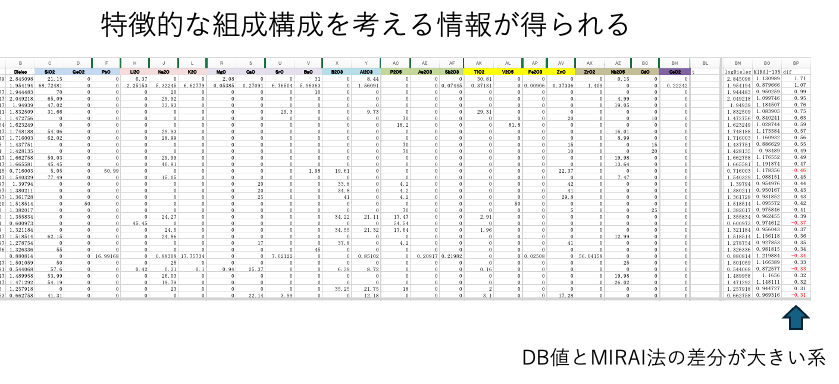
DB値とMIRAI法計算値の差分を大きい順にソートする。その時に組成の特徴が得られる。例えば、SiO2/Na2O/Nb2O5はMIRAI法計算値が小さくなる。
何故推算式を構築するのか、思い出す必要がある。
常に相関係数1.0のモデルができるなら、逆設計も簡単だ。量子コンピュータが実用化すれば組成のバラエティーが無限近くあっても最適値を探し出す事ができるだろう。
61種類の原料を1%刻みで全ての組み合わせを評価する。たかだか、(100)60計算すればいい。
それまでは、クロスタームや、MIRAI法の導き出す全体感をもとに、効率的な逆探索法を考えるしかない。その時には精度の高い推算式よりは、何が起きているか知見を与えてくれる推算式の方が価値は高い。
そこを勘違いしてあくせく相関係数の向上を目指して研究を行っている場合が多い。(自戒をこめて)
重回帰法の欠点を解消するLASSDGE法の効果をガラスの誘電率推算法を例に解説した[*8]。
[7. Pirika内リンク]
*1 拡張Appen式によるガラス物性推算法
*2 再構築学習法ニューラルネットワーク
*3 クラッシクなニューラルネットワーク法
*4 過学習と予測性の欠如
*5 重回帰法
*6 クロスターム重回帰法
*7 MIRAI法
*8 LASSDGE法
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)
メールの件名は[pirika]で始め